")
In January 2024, the American Society of Clinical Oncology (ASCO) in conjunction with the Society of Surgical Oncology released new guidelines for germline testing in patients with breast cancer, which include the following: For a full list of recommendations in this guideline, the article is available at: https://ascopubs.org/doi/10.1200/JCO.23.02225 Bedrosian, et al. J Clin Oncol. 2024;42(5):584-604. PMID:38175972. Social …
Tag: TP53
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2024-new-guidelines-released-through-asco-society-of-oncology-germline-testing-in-patients-with-breast-center/
ICARE Newsletter Spring 2024
National Comprehensive Cancer Network (NCCN) Guideline Updates
ICARE Newsletter Spring 2024
National Comprehensive Cancer Network (NCCN) Guideline Updates
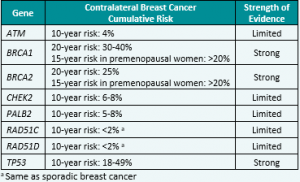
Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer – Released February 12th, 2024 (V3.2024) Check out the full guidelines by creating a FREE account at www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf Contralateral breast cancer risks in these updated guidelines: Expanded guidance about gynecologic cancers in BRCA1/2 carriers: Some highlights related to HRT include: Genetic/Familial High-Risk Assessment: Colorectal Cancer – Released …
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2024-national-comprehensive-cancer-network-nccn-guideline-updates/
ICARE Social Media Post January 2023
New ASCO Germline Testing in Patients With Breast Cancer Guidelines
ICARE Social Media Post January 2023
New ASCO Germline Testing in Patients With Breast Cancer Guidelines

The American Society of Clinical Oncology (ASCO) just released new guidelines for germline testing in patients with breast cancer, which include the following:🧬 BRCA1/2 testing should be offered to ALL patients diagnosed with breast cancer at or below age 65🧬 Testing for other hereditary cancer genes should also be offered based on personal and family …
Permanent link to this article: https://inheritedcancer.net/post10524/
ICARE Newsletter Fall 2023
National Comprehensive Cancer Network (NCCN) Guidelines Updates
ICARE Newsletter Fall 2023
National Comprehensive Cancer Network (NCCN) Guidelines Updates
Check out the full NCCN guidelines by creating a FREE account at www.nccn.org Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic CancerReleased August 28th, 2023 (V1.2024) › Transgender, Non-Binary, and Gender Diverse Individuals: NEW section on care (Page 63-66, TNBGD-1 to 4)› Li-Fraumeni Syndrome: Significant updates to content (risks and care) (Pages 57-60, LIFR-A): Table added …
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-national-comprehensive-cancer-network-nccn-guidelines-updates/
ICARE Social Media Post October 2023
What Are Established Cancer Genes?
ICARE Social Media Post October 2023
What Are Established Cancer Genes?

There are many different inherited breast cancer genes, and different genes lead to different levels of breast cancer risk:
Permanent link to this article: https://inheritedcancer.net/post100423/
ICARE Social Media Post June 2023
Li-Fraumeni Syndrome/TP53 mutations
ICARE Social Media Post June 2023
Li-Fraumeni Syndrome/TP53 mutations

A recent study found that, regardless of a prior cancer diagnosis, there was no increased cancer worry after 1 year of full-body MRI surveillance in TP53 carriers. Further research is needed to evaluate these issues further. Use the link in bio to read the full article! Reference: Omran, et al. Cancer. 2023;129(6):946-955. PMID: 36601958.
Permanent link to this article: https://inheritedcancer.net/post62123/
ICARE Featured Video June 2023
CHIP/Variant Interpretation in Li-Fraumeni Syndrome
ICARE Featured Video June 2023
CHIP/Variant Interpretation in Li-Fraumeni Syndrome
Below you may watch a featured video from the June 2023 Genetics Case Conference, which focused on CHIP and variant interpretation in Li-Fraumeni Syndrome with guest expert Bita Nehoray, MS, CGC from the City of Hope.
Permanent link to this article: https://inheritedcancer.net/video60823/
ICARE Social Media Post June 2023
Updates to NCCN Guidelines: Genetic/Familial High-Risk Assessment: Colorectal Version 1.2023
ICARE Social Media Post June 2023
Updates to NCCN Guidelines: Genetic/Familial High-Risk Assessment: Colorectal Version 1.2023

The National Comprehensive Cancer Network (NCCN) just released updated colorectal cancer guidelines which includes: You can check out the full guidelines by creating a FREE account at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf
Permanent link to this article: https://inheritedcancer.net/post60823/
ICARE Social Media Post March 2023
BGREAT December 2022 Newsletter
ICARE Social Media Post March 2023
BGREAT December 2022 Newsletter

Check out the latest edition of our B-GREAT newsletter for updates about inherited cancers in the context of racial inequalities in healthcare. You can read the newsletter by visiting 👇https://bgreatinitiative.inheritedcancer.net/wp-content/uploads/BGREAT-December-2022-Newsletter.pdf Please feel free to share with family members, friends, and/or your healthcare providers.
Permanent link to this article: https://inheritedcancer.net/post30623/
ICARE Social Media Post March 2023
Melanoma in TP53 /Li-Fraumeni Syndrome
ICARE Social Media Post March 2023
Melanoma in TP53 /Li-Fraumeni Syndrome
A new study found a 7-fold increased incidence of melanoma in individuals with TP53/Li-Fraumeni Syndrome compared with the general population:• Average age was 42• Risk to age 40 was 2.8%• Risk to age 60 was 7.8%• Risk to age 70 was 14.9% Learn more at the link below!https://pubmed.ncbi.nlm.nih.gov/35183552/ Reference: Hatton, et al. J Invest Dermatol. …
Permanent link to this article: https://inheritedcancer.net/post30723/
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-ask-the-expert/
ICARE Newsletter Spring 2022
Li-Fraumeni Syndrome and Cancer Risks
ICARE Newsletter Spring 2022
Li-Fraumeni Syndrome and Cancer Risks
A new study reported on differences in the TP53 mutations between patients who met vs those who did not meet Li-Fraumeni Syndrome (LFS) testing criteria. Several variants were identified multiple times in those who did and did not meet LFS clinical criteria: p.R175, p.G245, p.R248, p.R273, and p.R282. Other variants were exclusively found in those …
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-li-fraumeni-syndrome-and-cancer-risks/
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2022-which-genes-are-confirmed-as-inherited-breast-cancer-genes/
ICARE Featured Video August 2022
Total Body MRI Screening Protocol in Li-Fraumeni Syndrome Patients
ICARE Featured Video August 2022
Total Body MRI Screening Protocol in Li-Fraumeni Syndrome Patients
Below you may watch a featured video from the August 2022 Genetics Case Conference focused on total body MRI screening protocol in Li-Fraumeni Syndrome patients with guest expert Joanna Shechtel, MD.
Permanent link to this article: https://inheritedcancer.net/video81122/
ICARE Social Media Post May 2022
Breast Cancer Genes in Women of African Ancestry
ICARE Social Media Post May 2022
Breast Cancer Genes in Women of African Ancestry

A recent study in women of African ancestry confirmed genes previously identified to have associations with breast cancer risk (BRCA1, BRCA2, PALB2, ATM, TP53, NF1, and CHEK2) and provided new evidence of breast cancer risk for RAD51C and RAD51D, which was identified previously in European ancestry populations.Check out the full article at 👇https://pubmed.ncbi.nlm.nih.gov/35396981/Reference: Díaz-Zabala, et …
Permanent link to this article: https://inheritedcancer.net/post51722/
ICARE Social Media Post February 2022
Breast Cancer Characteristics Across Genes
ICARE Social Media Post February 2022
Breast Cancer Characteristics Across Genes

A new study demonstrated that breast cancer pathology and other clinical features differ by inherited breast cancer gene. Read the full article to learn more!https://jamanetwork.com/journals/jamaoncology/fullarticle/2788577Reference: Breast Cancer Association Consortium. JAMA Oncol. 2022 Jan 27. PMID: 35084436
Permanent link to this article: https://inheritedcancer.net/post20122/
ICARE Social Media Post December 2021
Li-Fraumeni Syndrome Cancer Risks
ICARE Social Media Post December 2021
Li-Fraumeni Syndrome Cancer Risks

• Several variants were identified multiple times in those who did and did not meet Li-Fraumeni Syndrome clinical criteria: p.R175, p.G245, p.R248, p.R273, and p.R282• Other variants were exclusively found in those in the Li-Fraumeni Syndrome group: p.M133T, p.P152L, p.C275Y, p.C275, p.R337C, p.R342, and p.R342P• One variant was exclusively found in patients with attenuated Li-Fraumeni …
Permanent link to this article: https://inheritedcancer.net/post122121/
ICARE Social Media Post August 2021
Metastatic Breast Cancer Genes
ICARE Social Media Post August 2021
Metastatic Breast Cancer Genes
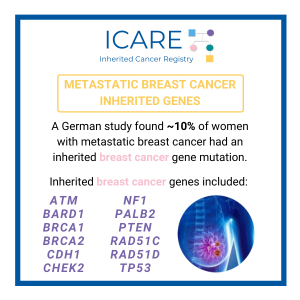
For additional information, read the Journal of Clinical Oncology article at the link below 👇 https://ascopubs.org/doi/10.1200/JCO.20.01200
Permanent link to this article: https://inheritedcancer.net/post82021/
ICARE Social Media Post November 2020
ASCO Guideline Updates: Breast Cancer
ICARE Social Media Post November 2020
ASCO Guideline Updates: Breast Cancer

The American Society of Clinical Oncology (ASCO) published updated guidelines for the management of hereditary breast cancer for the following gene carriers: 𝘽𝙍𝘾𝘼1/2 • Consider breast-conserving therapy • Consider nipple-sparing mastectomy, if medically appropriate • Advanced breast cancer: ⫸ PARP inhibitors (olaparib, talazoparib) preferred over non-platinum single agent chemotherapy ⫸ Platinum agents are recommended …
Permanent link to this article: https://inheritedcancer.net/post112020/
ICARE Social Media Post October 2020
TP53: Cancer Risks and Risk Management
ICARE Social Media Post October 2020
TP53: Cancer Risks and Risk Management

Gene: 𝙏𝙋𝟱𝟯 Syndrome: Li-Fraumeni Cancer Risks and Management per National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Breast/Ovarian/Pancreatic Version 1.2021: 𝗪𝗼𝗺𝗲𝗻: Breast cancer risk: Elevated at 54% – Recommend clinical breast exam every 6-12 months starting at age 20, annual breast MRI with contrast starting at age 20, and annual mammogram starting at age 30; …
Permanent link to this article: https://inheritedcancer.net/post101620/
ICARE Publication September 2020
Strategies to enhance identification of hereditary breast cancer gene carriers
ICARE Publication September 2020
Strategies to enhance identification of hereditary breast cancer gene carriers
No abstract available Reid S, et al. Strategies to enhance identification of hereditary breast cancer gene carriers. Expert Rev Mol Diagn. 2020 Sep; 20(9):861-865. Epub 2020 Sep 11. PMID: 32856489.
Permanent link to this article: https://inheritedcancer.net/pub91120/
ICARE Social Media Post July 2020
BRCA1/2 and Other Gene Carriers with Breast Cancer Don’t Always Receive Recommended Treatment
ICARE Social Media Post July 2020
BRCA1/2 and Other Gene Carriers with Breast Cancer Don’t Always Receive Recommended Treatment

BRCA1/2 and other gene mutation carriers with early stage breast cancer are not always receiving cancer treatment as recommended by national guidelines. Even though more and more people have been tested for hereditary cancer over the years, using this information accurately to guide treatment has not been as successful. These findings highlight the need for …
Permanent link to this article: https://inheritedcancer.net/post71020/
ICARE Newsletter Winter 2020
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic
ICARE Newsletter Winter 2020
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic

There were significant updates and restructuring of the guidelines, with some highlights included below: Substantial reorganization of the guidelines as follows: Now organized by organ site, rather than primarily by certain high penetrance genes Focused efforts to simplify genetic testing criteria Only one flow diagram included, to outline the ‘genetic testing process’ Following scenarios now …
Permanent link to this article: https://inheritedcancer.net/1nlw2020/
ICARE Social Media Post December 2019
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial Breast, Ovarian, and Pancreatic Guidelines (V1.2020)
ICARE Social Media Post December 2019
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial Breast, Ovarian, and Pancreatic Guidelines (V1.2020)

We are excited to share the latest version of the NCCN Genetic/Familial Breast, Ovarian and Pancreatic Guidelines (V1.2020), which were just updated. Some of the changes made include: PALB2 was added as a high penetrance gene (similar to BRCA1, BRCA2, CDH1, PTEN and TP53) It is appropriate to consider risk reducing mastectomy for cancer risk management …
Permanent link to this article: https://inheritedcancer.net/post12419/
ICARE Newsletter Summer 2019
Community Spotlight
ICARE Newsletter Summer 2019
Community Spotlight

Life was great at 45. I had nothing more than a few headaches and was a tad overweight. After a friend was diagnosed with breast cancer, I realized I had not had a mammogram in a couple of years, so I scheduled an appointment. One mass was found, but it was benign and nothing to …
Permanent link to this article: https://inheritedcancer.net/spotlightnls2019/
ICARE Newsletter Summer 2019
Expanding Our Thinking About Cancer Risks in TP53 Mutations and Li-Fraumeni Syndrome
ICARE Newsletter Summer 2019
Expanding Our Thinking About Cancer Risks in TP53 Mutations and Li-Fraumeni Syndrome

Since expanded genetic testing has become available through multigene panel tests, studies have suggested that many people identified to have TP53 mutations do not have a typical personal or family history, which is usually seen with Li-Fraumeni syndrome (LFS). A recent study looking at over 300 individuals with TP53 mutations (identified through multi-gene panel testing) …
Permanent link to this article: https://inheritedcancer.net/11nls2019/
ICARE Newsletter Summer 2018
Inherited Leukemias: The Importance of TP53/Li-Fraumeni Syndrome and Other Genes
ICARE Newsletter Summer 2018
Inherited Leukemias: The Importance of TP53/Li-Fraumeni Syndrome and Other Genes
It has long been established that the risk for developing leukemia in childhood is high among individuals with Li-Fraumeni Syndrome; however, better understanding the characteristics of leukemia among these individuals is important to guide treatment approaches. In a study of children with Acute Lymphocytic Leukemia (ALL), those with a germline TP53 mutation (compared to those …
Permanent link to this article: https://inheritedcancer.net/11nls2018/
ICARE Newsletter Summer 2018
New Data to Suggest Additional Genes Associated with Breast and Ovarian Cancer
ICARE Newsletter Summer 2018
New Data to Suggest Additional Genes Associated with Breast and Ovarian Cancer
A recent study reported on cancer risks among over 10,000 cancer patients across the United States who had genetic testing. Findings suggest breast cancer risks were associated with ATM, CHEK2, and PALB2, as expected; but an association was also found with MSH6 (in line with other recently published data, as outlined in another article in …
Permanent link to this article: https://inheritedcancer.net/7nls2018/
ICARE Newsletter Winter 2018
Updates to NCCN Genetic/Familial High-Risk Assessment: Breast and Ovarian Guidelines
ICARE Newsletter Winter 2018
Updates to NCCN Genetic/Familial High-Risk Assessment: Breast and Ovarian Guidelines
(Version 1.2018, posted Oct. 3, 2017) Metastatic prostate cancer was added as an indication for evaluation and testing for the BRCA1 and BRCA2 genes Among BRCA1, BRCA2, TP53 and PTEN carriers, women between ages 25-29 may consider having an annual mammogram with consideration of tomosynthesis if a breast MRI is not available. Among female BRCA2 …
Permanent link to this article: https://inheritedcancer.net/1nlw2018/
ICARE Newsletter Winter 2018
Study Suggests Inherited Cancer Genes Are Important in Pancreatic Cancer
ICARE Newsletter Winter 2018
Study Suggests Inherited Cancer Genes Are Important in Pancreatic Cancer

In a recent study which included over 800 patients with pancreatic ductal cancer, inherited cancer gene mutations were found in a much higher proportion than expected. Almost 5% of these patients had mutations identified in inherited cancer genes, the majority of which were in genes thought to be associated with pancreatic cancer (including BRCA2, ATM, …
Permanent link to this article: https://inheritedcancer.net/9nlw2018/
ICARE Newsletter Winter 2018
Advances in Cancer Screening Among Li-Fraumeni Syndrome Patients
ICARE Newsletter Winter 2018
Advances in Cancer Screening Among Li-Fraumeni Syndrome Patients
Several research groups from around the world that have conducted cancer screening among patients with Li-Fraumeni syndrome and a germline TP53 mutation have recently reported on their observations. Specifically, the National Cancer Institute group demonstrated that screening inclusive of rapid total body MRI detected cancers at an early stage,1 similar to findings published through other …
Permanent link to this article: https://inheritedcancer.net/5nlw2018/
ICARE Newsletter Summer 2017
What Are New and Subsequent Cancer Risks Among Patients with Li-Fraumeni Syndrome?
ICARE Newsletter Summer 2017
What Are New and Subsequent Cancer Risks Among Patients with Li-Fraumeni Syndrome?
Although individuals with Li-Fraumeni Syndrome (LFS), due to mutations in the TP53 gene, have a very high lifetime risk of cancer, risks of initial and subsequent cancers are not well defined. Through a group of patients with the classic form of LFS, researchers at the National Cancer Institute estimated their cancer risks. They evaluated a …
Permanent link to this article: https://inheritedcancer.net/3nls2017/
Permanent link to this article: https://inheritedcancer.net/2nls2017/
ICARE Newsletter Summer 2016
Surveillance Among Individuals with Li-Fraumeni Syndrome (LFS): An 11 Year Follow-Up Study
ICARE Newsletter Summer 2016
Surveillance Among Individuals with Li-Fraumeni Syndrome (LFS): An 11 Year Follow-Up Study
Results from the original screening protocol for LFS1 were recently updated following collection of 11 years of follow-up data.2 Through this study, 89 patients with LFS were given the option of a clinical surveillance protocol consisting of a physical examination as well as frequent biochemical and imaging studies. Forty asymptomatic tumors were detected in 32% …
Permanent link to this article: https://inheritedcancer.net/4nls2016/
ICARE Newsletter Winter 2016
Improving Our Understanding of Cancer Risks Among Individuals with Li-Fraumeni Syndrome
ICARE Newsletter Winter 2016
Improving Our Understanding of Cancer Risks Among Individuals with Li-Fraumeni Syndrome
A recent study from France included over 400 patients with Li-Fraumeni Syndrome (all of whom had an inherited TP53 gene mutation). Cancer types among children and adults differed, with the main cancer types among children being osteosarcomas, adrenocortical carcinomas, central nervous system (CNS) tumors and soft tissue sarcomas; whereas among adults, the main cancer types …
Permanent link to this article: https://inheritedcancer.net/5nlw2016/
Permanent link to this article: https://inheritedcancer.net/1nls2015/
Follow us to stay informed
Click below to read recent updates on inherited cancers

Click below to read inspiring stories from our ICARE participants
