")
Select updates outlined below. Check out the full guidelines by creating a FREE account at https://www.nccn.org/guidelines/category_2 Colorectal, Endometrial, & Gastric Cancer V1.2025 – Released June 13th, 2025 Breast, Ovarian, Pancreatic, & Prostate Cancer V1.2026 – Released July 10th, 2025
Tag: MSH2
Permanent link to this article: https://inheritedcancer.net/nlf20251/
ICARE Newsletter Fall 2025
Ovarian Cancer: 1 in 4 Cases Could Have Been Prevented!
ICARE Newsletter Fall 2025
Ovarian Cancer: 1 in 4 Cases Could Have Been Prevented!
A new study of 1877 ovarian cancer patients showed almost 25% of patients had ‘missed opportunities’ for salpingectomy (removal of fallopian tubes) when they had another surgery or procedure before their ovarian cancer diagnosis. Additionally, 6% of patients had a close relative with ovarian cancer, and almost 20% had a mutation in an ovarian cancer …
Permanent link to this article: https://inheritedcancer.net/nlf20253/
ICARE Social Media Post August 2025
NCCN CEG Guidelines (V1.2025): MSH2 Post
ICARE Social Media Post August 2025
NCCN CEG Guidelines (V1.2025): MSH2 Post

The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Gastric Cancer guidelines on June 13th, 2025 (Version 1.2025). Included in these new guidelines are updates to the MSH2 content. To read more, check out the full guidelines by creating a FREE account at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_ceg.pdf
Permanent link to this article: https://inheritedcancer.net/post08272025/
ICARE Social Media Post Month Year
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update (V1.2025)
ICARE Social Media Post Month Year
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update (V1.2025)

The National Comprehensive Cancer Network (NCCN) just released updated Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Gastric Cancer guidelines on June 13th, 2025 (Version 1.2025). There are many updates found in these new guidelines including updates to MSH2, EPCAM, PMS2, CDH1, NTHL1, and TP53 content as well as updates to the clinical diagnostic criteria for Peutz-Jeghers Syndrome. …
Permanent link to this article: https://inheritedcancer.net/post071425/
ICARE Social Media Post June 2025
ATM, CHEK2, & PALB2 Carriers: Are There Differences in Cancer-associated Mortality?
ICARE Social Media Post June 2025
ATM, CHEK2, & PALB2 Carriers: Are There Differences in Cancer-associated Mortality?

A new study showed that compared to non-carriers, ATM, CHEK2, and PALB2 carriers showed similar mortality from breast cancer, pancreatic cancer, and colorectal cancer. Other findings among BRCA1/2 carriers and Lynch Syndrome patients showed: Why is this important?These results may be reassuring for ATM, CHEK2, and PALB2 carriers, and provide additional useful information when discussing …
Permanent link to this article: https://inheritedcancer.net/post060625/
ICARE Social Media Post June 2025 2025
PREMM5: Model to Estimate the Risk for Having Lynch Syndrome
ICARE Social Media Post June 2025 2025
PREMM5: Model to Estimate the Risk for Having Lynch Syndrome

PREMM5 is a model to estimate the risk for having Lynch Syndrome. PREMMplus is a model that estimates risks in 19-cancer risk genes, including Lynch Syndrome genes, BRCA, and other genes. A new study that compared PREMM5 and PREMMplus found that PREMMplus was just as good as PREMM5 in identifying patients with Lynch Syndrome. PREMMplus …
Permanent link to this article: https://inheritedcancer.net/post060325/
ICARE Newsletter Spring 2025
Community Spotlight
ICARE Newsletter Spring 2025
Community Spotlight

As someone with two inherited cancer gene mutations—MSH6 (Lynch syndrome) and CHEK2 — I know firsthand the emotional and practical complexities of navigating hereditary cancer risk. My journey began without what many might consider “classic” red flags — just a few scattered cancer cases in my family, none of which seemed connected at the time. …
Permanent link to this article: https://inheritedcancer.net/12nls2025/
ICARE Newsletter Spring 2025
Lynch Syndrome: Personalizing Risks
ICARE Newsletter Spring 2025
Lynch Syndrome: Personalizing Risks

MyLynch is a resource for Lynch syndrome patients that provides personal cancer risks, education on interventions, and adjusted risk estimates, depending on the intervention(s) the patient chooses to pursue. If you have Lynch syndrome, go to https://hereditarycancer.dfci.harvard.edu/mylynch/ to get your personalized risk estimate. Check out a recent presentation by Dr. Yurgelun, who helped develop MyLynch, …
Permanent link to this article: https://inheritedcancer.net/6nls2025/
ICARE Newsletter Spring 2025
National Comprehensive Cancer Network (NCCN) Guideline Updates
ICARE Newsletter Spring 2025
National Comprehensive Cancer Network (NCCN) Guideline Updates
Breast, Ovarian, Pancreatic, and Prostate Cancer Colorectal, Endometrial, and Gastric Cancer
Permanent link to this article: https://inheritedcancer.net/1nls2025/
ICARE Social Media Post October 2024
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update V2.2024 – #2 EPCAM Updates
ICARE Social Media Post October 2024
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update V2.2024 – #2 EPCAM Updates

The National Comprehensive Cancer Network (NCCN) released updated Genetic Familial High-Risk Assessment Colorectal, Endometrial, and Gastric Cancer guidelines on October 3rd, 2024. In these updated guidelines, NCCN revised information about EPCAM gene (which has usually been lumped together with MSH2) as follows ⤸ You can check out the full guidelines by creating a FREE account …
Permanent link to this article: https://inheritedcancer.net/post102224/
ICARE Social Media Post October 2024
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update V2.2024
ICARE Social Media Post October 2024
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update V2.2024

The National Comprehensive Cancer Network (NCCN) released updated Genetic Familial High-Risk Assessment Colorectal, Endometrial, and Gastric Cancer guidelines on October 3rd, 2024. Updates include ⤸ Added the following to testing being considered: Personal history of colorectal or endometrial cancer at or older than age 50, and: Revised information about EPCAM gene (which has usually been …
Permanent link to this article: https://inheritedcancer.net/post100824/
ICARE Newsletter Spring 2024
National Comprehensive Cancer Network (NCCN) Guideline Updates
ICARE Newsletter Spring 2024
National Comprehensive Cancer Network (NCCN) Guideline Updates
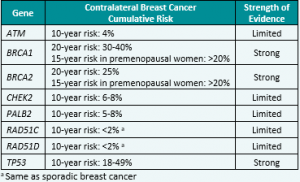
Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer – Released February 12th, 2024 (V3.2024) Check out the full guidelines by creating a FREE account at www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf Contralateral breast cancer risks in these updated guidelines: Expanded guidance about gynecologic cancers in BRCA1/2 carriers: Some highlights related to HRT include: Genetic/Familial High-Risk Assessment: Colorectal Cancer – Released …
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2024-national-comprehensive-cancer-network-nccn-guideline-updates/
ICARE Social Media Post January 2023
Colon Adenomas in Lynch Syndrome
ICARE Social Media Post January 2023
Colon Adenomas in Lynch Syndrome
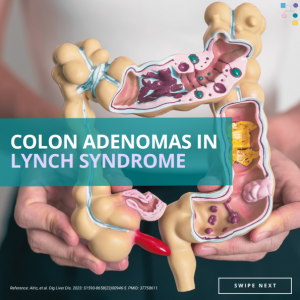
New research reveals that individuals under the age of 50 with Lynch Syndrome often develop small, flat adenomas, particularly in the right (proximal) colon. This finding emphasizes the importance of rigorous screening, with a special focus on the proximal colon, among these young Lynch Syndrome patients 🩺🔍 Learn more at: https://www.dldjournalonline.com/article/S1590-8658(23)00946-5/fulltext Reference: Alric, et al. …
Permanent link to this article: https://inheritedcancer.net/post11024/
ICARE Newsletter Fall 2023
Genes Associated with Aggressive Prostate Cancer
ICARE Newsletter Fall 2023
Genes Associated with Aggressive Prostate Cancer
A new study of almost 18,000 men with prostate cancer showed that inherited mutations in the BRCA2, ATM, and NBN genes were strongly associated with aggressive prostate cancer. Less strong associations were seen for inherited mutations in the MSH2, XRCC2, and MRE11A genes. The findings of this study suggest that knowing about inherited genes that …
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-genes-associated-with-aggressive-prostate-cancer/
ICARE Newsletter Fall 2023
National Comprehensive Cancer Network (NCCN) Guidelines Updates
ICARE Newsletter Fall 2023
National Comprehensive Cancer Network (NCCN) Guidelines Updates
Check out the full NCCN guidelines by creating a FREE account at www.nccn.org Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic CancerReleased August 28th, 2023 (V1.2024) › Transgender, Non-Binary, and Gender Diverse Individuals: NEW section on care (Page 63-66, TNBGD-1 to 4)› Li-Fraumeni Syndrome: Significant updates to content (risks and care) (Pages 57-60, LIFR-A): Table added …
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-national-comprehensive-cancer-network-nccn-guidelines-updates/
ICARE Social Media Post June 2023
Updates to NCCN Guidelines: Genetic/Familial High-Risk Assessment: Colorectal Version 1.2023
ICARE Social Media Post June 2023
Updates to NCCN Guidelines: Genetic/Familial High-Risk Assessment: Colorectal Version 1.2023

The National Comprehensive Cancer Network (NCCN) just released updated colorectal cancer guidelines which includes: You can check out the full guidelines by creating a FREE account at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf
Permanent link to this article: https://inheritedcancer.net/post60823/
ICARE Newsletter Spring 2023
Lynch Syndrome: Colorectal Cancer Risks Revisited
ICARE Newsletter Spring 2023
A study of 381 individuals with Lynch Syndrome in New Zealand (98 with Lynch Syndrome-associated variants in MLH1, 159 in MSH2, 103 in MSH6, and 21 in PMS2) found that risks for colorectal cancer were lower in MSH6 and PMS2 carriers, suggesting that it might be possible to spread out colonoscopy intervals for these individuals.1 …
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2023/
ICARE Social Media Post March 2023
Metachronous Colorectal Cancer in Lynch Syndrome
ICARE Social Media Post March 2023
Metachronous Colorectal Cancer in Lynch Syndrome

Findings from a recent study: Risk factors for metachronous colorectal cancer included a history of colorectal polyps and having an MLH1 or MSH2 mutation, while protective factors included female sex and extended surgical resection. This highlights the importance of genetic testing and counseling for Lynch syndrome prior to surgery, which can influence surgical strategy and …
Permanent link to this article: https://inheritedcancer.net/post31923/
ICARE Newsletter Spring 2022
Lynch Syndrome and Prostate Cancer
ICARE Newsletter Spring 2022
Lynch Syndrome and Prostate Cancer
A new study suggests men with certain MSH2 and MSH6 mutations have higher risks of prostate cancer and may be candidates for PSA screening. Bancroft et al. Lancet Oncol. 2021 Nov. PMID: 34678156. Social media postDecember 17th, 2021. Available at: https://tinyurl.com/post121721
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-lynch-syndrome-and-prostate-cancer/
Newsletter Fall 2021
Updates to NCCN Genetic/Familial High-Risk Assessment
Newsletter Fall 2021
Updates to NCCN Genetic/Familial High-Risk Assessment
Breast, Ovarian, and Pancreatic Guidelines V.1.2022: Released August 11th, 2021 Colorectal Cancer Guidelines V.1.2021: Released May 11th, 2021 Check out the full NCCN guidelines by creating a FREE account at www.nccn.org
Permanent link to this article: https://inheritedcancer.net/newsletter-fall-2021-updates-to-nccn-genetic-familial-high-risk-assessment/
ICARE Social Media Post July 2022
Pancreatic Cancer Screening
ICARE Social Media Post July 2022
Pancreatic Cancer Screening

A recent study found that earlier diagnosis improved survival in people at high risk of pancreatic cancer.High risk was defined based on:family history and/orinherited gene mutation (BRCA1, BRCA2, CDKN2A, Lynch Syndrome genes, PALB2, ATM, and STK11)Read the article at the link: https://ascopost.com/news/june-2022/outcomes-of-pancreas-surveillance-in-the-caps5-study-and-total-caps-cohort/Reference: Dbouk, et al. J Clin Oncol. 2022 Jun 15:JCO2200298. doi: 10.1200/JCO.22.00298. PMID: 35704792.
Permanent link to this article: https://inheritedcancer.net/post72622/
ICARE Social Media Post December 2021
Prostate Cancer Screening: MSH2 & MSH6
ICARE Social Media Post December 2021
Prostate Cancer Screening: MSH2 & MSH6

A new study suggests men with MSH2 and MSH6 mutations have a higher incidence of prostate cancer, and may be candidates for PSA screening. Read the full article to learn more 👇 https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(21)00522-2/fulltext Reference: Bancroft et al. Lancet Oncol. 2021 Nov;22(11):1618-1631. PMID: 34678156.
Permanent link to this article: https://inheritedcancer.net/post121721/
ICARE Social Media Post November 2021
Variation in Colorectal Cancer Risk in Families With Lynch Syndrome
ICARE Social Media Post November 2021
Variation in Colorectal Cancer Risk in Families With Lynch Syndrome

For more information, read the Lancet Oncology article led by The International Mismatch Repair Consortium at the below link 👇https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(21)00189-3/fulltext Reference: International Mismatch Repair Consortium. Lancet Oncol. 2021 Jul;22(7):1014-1022. PMID: 34111421.
Permanent link to this article: https://inheritedcancer.net/post113021/
ICARE Social Media Post March 2021
Winter 2021 Community Spotlight
ICARE Social Media Post March 2021
Winter 2021 Community Spotlight

In every ICARE newsletter we feature a 𝗰𝗼𝗺𝗺𝘂𝗻𝗶𝘁𝘆 𝘀𝗽𝗼𝘁𝗹𝗶𝗴𝗵𝘁 to share their experience with inherited cancer. In the latest edition, Dave Dubin, co-founder of AliveAndKickn, shares his experience with Lynch Syndrome. Check out his full story at: https://inheritedcancer.net/community-spotlight/
Permanent link to this article: https://inheritedcancer.net/post31921/
ICARE Featured Video March 2021
Inherited Colorectal Cancer
ICARE Featured Video March 2021
Inherited Colorectal Cancer

Below you may watch a featured video from the March 2021 Genetics Case Conference, which focused on inherited colorectal cancer with guest expert Georgia Wiesner, MD from Vanderbilt University Medical Center.
Permanent link to this article: https://inheritedcancer.net/video31121/
ICARE Social Media Post December 2020
Broader Germline Testing for Urothelial Cancer
ICARE Social Media Post December 2020
Broader Germline Testing for Urothelial Cancer

The most common inherited form of urothelial cancers is Lynch Syndrome. However, a study showed that of 586 individuals with urothelial cancer, 80 had a mutation in an inherited cancer gene (14%). Mutations in several genes were observed; however, 𝙈𝙎𝙃2 and 𝘽𝙍𝘾𝘼2 were both significantly associated with urothelial cancer (odds ratio of 3.7). Confirmatory …
Permanent link to this article: https://inheritedcancer.net/post120420/
ICARE Social Media Post September 2020
Living with Lynch 2020 Virtual Patient Workshop
ICARE Social Media Post September 2020
Living with Lynch 2020 Virtual Patient Workshop

SJoin the Colon Cancer Coalition and AliveAndKickn for the Living with Lynch 2020 Virtual Patient Workshop on𝗙𝗿𝗶𝗱𝗮𝘆, 𝗢𝗰𝘁𝗼𝗯𝗲𝗿 𝟵𝘁𝗵 𝗳𝗿𝗼𝗺 𝟭𝗽𝗺-𝟱𝗽𝗺 𝗘𝗧 to hear unique patient perspectives and the latest information from experts on Lynch syndrome. Visit https://www.livingwithlynch.org/2020-living-with-lynch to register for this free workshop today!
Permanent link to this article: https://inheritedcancer.net/post92920/
ICARE Newsletter Summer 2020
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal Guidelines
ICARE Newsletter Summer 2020
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal Guidelines

Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal Guidelines (Version 1.2020, posted July 21, 2020) For individuals with Lynch Syndrome: Cancer risks were updated based on information from recent studies: Main updates included cancer risks in PMS2 (endometrial, ovarian, and prostate cancer), MSH2 and EPCAM (prostate and brain cancer), and MSH6 (prostate …
Permanent link to this article: https://inheritedcancer.net/1nls2020/
ICARE Social Media Post July 2020
Updates to 2020 NCCN Genetic/Familial Colorectal Guidelines
ICARE Social Media Post July 2020
Updates to 2020 NCCN Genetic/Familial Colorectal Guidelines
The National Comprehensive Cancer Network (NCCN) released new guidelines for 2020 on July 21, 2020. The big changes included refining some of the risks for genes involved in Lynch Syndrome, and providing specific guidance about cancer screening that may slightly differ by gene. You can check out the full guidelines by creating a FREE account …
Permanent link to this article: https://inheritedcancer.net/post72420/
ICARE Social Media Post March 2020
MSH2: Cancer Risks and Risk Management
ICARE Social Media Post March 2020
MSH2: Cancer Risks and Risk Management

Gene: MSH2 Cancer Risks and Management (per NCCN version 3.2019): Women: Endometrial cancer risk: Elevated at 21%-57% – Consider risk-reducing hysterectomy. Ovarian cancer risk: Elevated at 10%-38% – Recommend risk-reducing bilateral salpingo-oophorectomy (removal of ovaries and fallopian tubes). Men and Women: Colorectal cancer risk: Elevated at 43%-52% – Recommend colonoscopy every 1-2 years starting at …
Permanent link to this article: https://inheritedcancer.net/post31020/
ICARE Newsletter Winter 2020
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic
ICARE Newsletter Winter 2020
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic

There were significant updates and restructuring of the guidelines, with some highlights included below: Substantial reorganization of the guidelines as follows: Now organized by organ site, rather than primarily by certain high penetrance genes Focused efforts to simplify genetic testing criteria Only one flow diagram included, to outline the ‘genetic testing process’ Following scenarios now …
Permanent link to this article: https://inheritedcancer.net/1nlw2020/
ICARE Newsletter Winter 2020
Updated Pancreatic Cancer Screening Guidelines through CAPS Consortium
ICARE Newsletter Winter 2020
Updated Pancreatic Cancer Screening Guidelines through CAPS Consortium

The International Cancer of the Pancreas Screening (CAPS) Consortium recently published updated recommendations about pancreatic cancer screening through MRI/magnetic retrograde cholangiopancreatography (MRCP) and/or an endoscopic ultrasound (EUS).1 Specifically, these guidelines now recommend that individuals with a CDKN2A or STK11 mutation begin screening at age 40. Screening for individuals with a BRCA1/2, ATM, PALB2, MLH1, or …
Permanent link to this article: https://inheritedcancer.net/4nlw2020/
ICARE Newsletter Winter 2020
Lynch Syndrome Cancer Risks Across Genes
ICARE Newsletter Winter 2020
Lynch Syndrome Cancer Risks Across Genes

A worldwide study reporting on cancer risks among individuals with mutations in Lynch syndrome genes showed that there are substantial differences in cancer risks across the various genes.1 Specifically, the risk for colorectal cancer in those with MLH1, MSH2, and MSH6 mutations was substantially higher than what was seen for those with PMS2 mutations. Additionally, …
Permanent link to this article: https://inheritedcancer.net/5nlw2020/
ICARE Social Media Post February 2020
PMS2 and MSH6 Colorectal Cancer Risks
ICARE Social Media Post February 2020
PMS2 and MSH6 Colorectal Cancer Risks

Individuals with Lynch syndrome have an increased risk of colorectal cancer (CRC) and other cancers. The level of CRC risk is different based on which gene they have a mutation in. Of note, MLH1 and MSH2 carriers have the highest risk of colorectal cancer, generally in the range of 43%-52% by age 70. A recent …
Permanent link to this article: https://inheritedcancer.net/post22020/
ICARE Social Media Post February 2020
Lynch Syndrome Cancer Risks Across Genes
ICARE Social Media Post February 2020
Lynch Syndrome Cancer Risks Across Genes

A worldwide study suggests that risks for cancers for the various Lynch syndrome genes have some differences. The risk of colorectal cancer for those with a mutation in the MLH1, MSH2 and MSH6 genes is higher than what is seen for carriers of a PMS2 mutation. Additionally, men with MSH2 gene mutations have a higher …
Permanent link to this article: https://inheritedcancer.net/post2720/
ICARE Social Media Post February 2020
Differences in Pancreatic Cancer Screening Recommendations from the National Comprehensive Cancer Network (NCCN) and the International Cancer of the Pancreas Screening (CAPS) Consortium
ICARE Social Media Post February 2020
Differences in Pancreatic Cancer Screening Recommendations from the National Comprehensive Cancer Network (NCCN) and the International Cancer of the Pancreas Screening (CAPS) Consortium
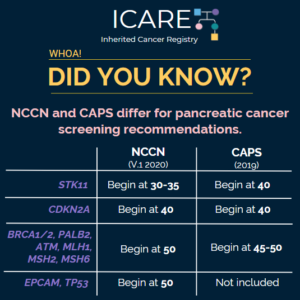
The National Comprehensive Cancer Network (NCCN) and the International Cancer of the Pancreas Screening (CAPS) Consortium recently updated pancreatic cancer screening recommendations. However, there are some differences between these recommendations. Specifically, screening with annual MRI/magnetic retrograde cholangiopancreatography (MRCP) and/or endoscopic ultrasound (EUS) is recommended as follows for NCCN versus CAPS: STK11 regardless of family history: …
Permanent link to this article: https://inheritedcancer.net/post2620/
ICARE Social Media Post February 2020
Updated Pancreatic Cancer Screening Guidelines through the International Cancer of the Pancreas Screening (CAPS) Consortium
ICARE Social Media Post February 2020
Updated Pancreatic Cancer Screening Guidelines through the International Cancer of the Pancreas Screening (CAPS) Consortium

The International Cancer of the Pancreas Screening (CAPS) Consortium recently published updated pancreatic cancer screening recommendations. The recommendations include: Screening with MRI/magnetic retrograde cholangiopancreaography (MRCP) and/or endoscopic ultrasound (EUS) The screening was recommended for the following individuals: CDKN2A and STK11 mutation carriers starting at age 40 BRCA1/2, ATM, PALB2, MLH1, and MSH2 mutation carriers (if …
Permanent link to this article: https://inheritedcancer.net/post2420/
ICARE Social Media Post December 2019
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial Breast, Ovarian, and Pancreatic Guidelines (V1.2020)
ICARE Social Media Post December 2019
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial Breast, Ovarian, and Pancreatic Guidelines (V1.2020)

We are excited to share the latest version of the NCCN Genetic/Familial Breast, Ovarian and Pancreatic Guidelines (V1.2020), which were just updated. Some of the changes made include: PALB2 was added as a high penetrance gene (similar to BRCA1, BRCA2, CDH1, PTEN and TP53) It is appropriate to consider risk reducing mastectomy for cancer risk management …
Permanent link to this article: https://inheritedcancer.net/post12419/
ICARE Newsletter Summer 2019
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal Guidelines
ICARE Newsletter Summer 2019
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal Guidelines

(Version 1.2019, posted July 3, 2019) For Individuals with Lynch Syndrome: The cancer risk table was updated: Addition of new cancer risks by specific genes: breast and bladder cancers Updates of cancer risks by specific genes: ovarian, prostate, gastric, pancreatic, urothelial, small bowel, and brain/CNS cancers Removal of reference to sebaceous neoplasms Recommendations for cancer …
Permanent link to this article: https://inheritedcancer.net/1nls2019/
ICARE Newsletter Winter 2019
Other Advances in Cancer Treatment Among Cancer Patients with Inherited Disease: Lynch Syndrome
ICARE Newsletter Winter 2019
Other Advances in Cancer Treatment Among Cancer Patients with Inherited Disease: Lynch Syndrome
Pertaining to metastatic prostate cancer, recently published data reported 8.1% of men with advanced prostate cancer had evidence of mismatch repair (MMR) mutations in their tumors. These types of mutations are frequently seen in tumors among Lynch syndrome patients. In addition, men with this type of tumor had much poorer survival. Tumors with MMR defects …
Permanent link to this article: https://inheritedcancer.net/7nlw2019/
ICARE Newsletter Winter 2019
Basal Cell Cancers May Be a Risk Factor to Predict Inherited Cancer Predisposition
ICARE Newsletter Winter 2019
Basal Cell Cancers May Be a Risk Factor to Predict Inherited Cancer Predisposition
An interesting area of progress to identify individuals with inherited risks included a study of over 13,000 individuals with six or more basal cell cancers (BCC) evaluated through a claims database. Results indicated ~20% of these individuals had a germline mutation in a DNA repair gene, including BRCA1/2, PALB2, and the Lynch syndrome genes, among …
Permanent link to this article: https://inheritedcancer.net/10nlw2019/
ICARE Newsletter Winter 2019
Expansion of Lynch Syndrome Tumor Spectrum Which May Have Treatment Implications
ICARE Newsletter Winter 2019
Expansion of Lynch Syndrome Tumor Spectrum Which May Have Treatment Implications
Although the Lynch syndrome tumor spectrum is thought to be limited to cancers of the colorectum, endometrium, ovaries, stomach, and a few other cancer types, a recent article suggested there might be a broader tumor spectrum than previously considered. Furthermore, colorectal and endometrial cancers which develop among Lynch syndrome patients frequently are determined on tumor …
Permanent link to this article: https://inheritedcancer.net/6nlw2019/
ICARE Newsletter Summer 2018
Differences in Breast Cancer Risks Among Women with Lynch Syndrome
ICARE Newsletter Summer 2018
Differences in Breast Cancer Risks Among Women with Lynch Syndrome
Breast cancer risks were recently reported among a sample of 423 women with mutations in one of the Lynch syndrome genes (MLH1, MSH2, MSH6, or PMS2).1 Results indicated that breast cancer risks were substantially higher among those with MSH6 and PMS2 mutations, compared to MLH1 and MSH2 mutations. In fact, breast cancer risk to age …
Permanent link to this article: https://inheritedcancer.net/3nls2018/
ICARE Newsletter Summer 2018
Refining Cancer Risks Among Individuals with Lynch Syndrome
ICARE Newsletter Summer 2018
Refining Cancer Risks Among Individuals with Lynch Syndrome
Over the past year, multiple studies have refined risks and types of cancer among individuals with Lynch syndrome. Through a Scandinavian study, risks for 13 types of cancer (with colorectal cancers being excluded), were reported to be elevated with differences related to gender, age, and the gene in which mutation was present. Incidence rates of …
Permanent link to this article: https://inheritedcancer.net/5nls2018/
ICARE Newsletter Winter 2018
Advances in New Treatments for Individuals with Lynch Syndrome
ICARE Newsletter Winter 2018
Advances in New Treatments for Individuals with Lynch Syndrome
A recently published phase II clinical trial investigated the use of a new class of drugs (called PD-1 Inhibitors) in DNA mismatch repair-deficient/ microsatellite instability-high colorectal tumors (which are features seen in the majority of colorectal tumors from individuals with Lynch Syndrome) among patients with metastatic disease.1 Investigators found patients who received two PD-1 Inhibitors …
Permanent link to this article: https://inheritedcancer.net/7nlw2018/
ICARE Newsletter Winter 2018
Study Suggests Inherited Cancer Genes Are Important in Pancreatic Cancer
ICARE Newsletter Winter 2018
Study Suggests Inherited Cancer Genes Are Important in Pancreatic Cancer

In a recent study which included over 800 patients with pancreatic ductal cancer, inherited cancer gene mutations were found in a much higher proportion than expected. Almost 5% of these patients had mutations identified in inherited cancer genes, the majority of which were in genes thought to be associated with pancreatic cancer (including BRCA2, ATM, …
Permanent link to this article: https://inheritedcancer.net/9nlw2018/
Permanent link to this article: https://inheritedcancer.net/2nls2017/
ICARE Newsletter Winter 2017
The Potential Promise of Immunotherapy Targeted to Those with Bi-Allelic Mutations in Lynch Syndrome Genes
ICARE Newsletter Winter 2017
The Potential Promise of Immunotherapy Targeted to Those with Bi-Allelic Mutations in Lynch Syndrome Genes
People with Lynch Syndrome have a non-working Lynch gene (“mutation”), while the other copy of that gene is normal (recognizing that all of these genes come in pairs, with one member of the pair coming from each parent). Over the last few years, there has been an increased realization that some individuals have a mutation …
Permanent link to this article: https://inheritedcancer.net/2nlw2017/
ICARE Newsletter Summer 2016
Practice Guideline Updates for NCCN Genetic/Familial High-Risk Assessment
ICARE Newsletter Summer 2016
Practice Guideline Updates for NCCN Genetic/Familial High-Risk Assessment
The National Comprehensive Cancer Network (NCCN) is a network of oncology healthcare providers who work together to develop best practice guidelines for the delivery of cancer care. Given the increasing use of testing for mutations in several inherited cancer genes at one time (called “multi-gene panel testing”), the Breast/Ovarian and Colorectal Panels sought to provide …
Permanent link to this article: https://inheritedcancer.net/2nls2016/
ICARE Newsletter Winter 2016
What Is the Risk for Ovarian Cancer Among Women with Mutations in Newer Ovarian Cancer Genes?
ICARE Newsletter Winter 2016
What Is the Risk for Ovarian Cancer Among Women with Mutations in Newer Ovarian Cancer Genes?
The most common form of inherited ovarian cancer is due to mutations in the BRCA1 and BRCA2 genes, which are present in 10-15% of women with ovarian cancer and lead to an ovarian cancer risk of up to 44% and 27%, respectively. Another set of genes known to raise ovarian cancer risks are the mismatch …
Permanent link to this article: https://inheritedcancer.net/2nlw2016/
ICARE Newsletter Summer 2014
Is There a Higher Risk of Prostate Cancer in Individuals with Lynch Syndrome?
ICARE Newsletter Summer 2014
Is There a Higher Risk of Prostate Cancer in Individuals with Lynch Syndrome?
Over the last few years, there have been studies to suggest that men with Lynch Syndrome may have a higher risk for developing prostate cancer.1,2,3,4,5 The results of these studies have differed as to whether there is an association with an aggressive form of disease. For example, some studies report the risk of developing prostate …
Permanent link to this article: https://inheritedcancer.net/6nls2014/
ICARE Newsletter Winter 2013
Is Lynch Syndrome Associated with Breast Cancer?
ICARE Newsletter Winter 2013
Is Lynch Syndrome Associated with Breast Cancer?
The cancer spectrum typically seen in individuals with Lynch Syndrome includes cancers of the colon, endometrium, ovary, stomach, and other cancers (including cancer of the renal pelvis, ureter, small bowel and pancreas). The issue of whether breast cancer risk is elevated in those with Lynch syndrome has been controversial, with conflicting results between various studies. …
Permanent link to this article: https://inheritedcancer.net/3nlw2013/


Click below to read recent updates on inherited cancers
Click below to read inspiring stories from our ICARE participants
