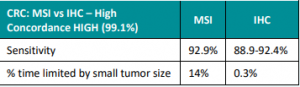
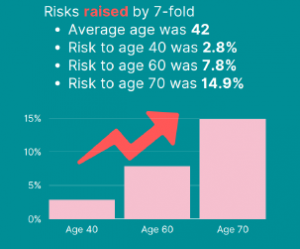
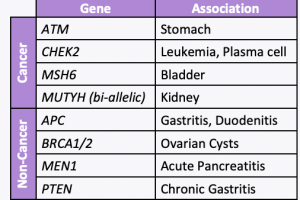
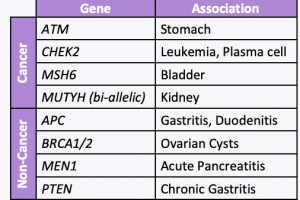
NCCN just released new Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Esophageal, and Gastric Cancer Guidelines (Version 1.2026) on June 16th, 2026! For Lynch Syndrome-associated cancers, genetic testing recommended for: For all other Lynch-associated cancers, tumor screening for MMR deficiency recommended regardless of age at diagnosis, including: Reference: Latham A, et al. J Clin Oncol 2019;37(4):286-295. PMID: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post062326/
ICARE Social Media Post June 2026
Esophageal Cancer Added to NCCN Guidelines (V1.2026)
The new NCCN Genetic/Familial High-Risk Assessment Guidelines Version 1.2026 (released June 16th, 2026) are now called: Colorectal, Endometrial, Esophageal, and Gastric Cancer Guidelines NOTE: Esophageal cancer newly added to name of this guideline! In these guidelines, an entire section was added on hereditary esophageal cancer (see pages HESOPH-1 and HESOPH-2:PRINCIPLES OF GENETIC RISK ASSESSMENT FOR …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post061826/
ICARE Social Media Post June 2026
NCCN Colorectal, Endometrial, Esophageal, and Gastric Cancer Guidelines (V1.2026)
New NCCN Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Esophageal, and Gastric Cancer Guidelines (Version 1.2026) were just released on June 16th, 2026! For the purpose of identifying Lynch syndrome, genetic testing is recommended for all patients with the following (regardless of age): There was a ‘category 2b’ distinction for the below scenarios: Category 2b means that …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post061726/
ICARE Social Media Post June 2026
NHS Galleri Trial: Blood Test for Cancer Detection
A new study (NHS-Galleri trial) tested whether a simple blood test could catch more cancers early. Bottom line: Stage 3 and 4 cancers (together) were not lowered by this study, so this test is NOT ready for prime time yet! A small silver lining is that when looking at stage 4 cancers alone, there was …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post061626/
ICARE Social Media Post June 2026
Young Black Women with Breast Cancer: Clinicopathologic Characteristics
A new study among Black women diagnosed with breast cancer at or below age 50 found that 15.3% had germline pathogenic/likely pathogenic variants in breast and/or ovarian cancer predisposition genes. 🧬The most common were BRCA1 (7.7%), BRCA2 (4.5%), PALB2 (0.9%), and ATM (0.7%)🧬When compared with sporadic cancers, BRCA1 carriers were far more likely to have …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post060926/
ICARE Social Media Post June 2026
BRCA1: Hormone Replacement Therapy After Ovary Removal
A new study shows that estrogen-only hormone replacement therapy (HRT) after ovary removal can LOWER breast cancer risk in women carrying the BRCA1 genetic mutation. What were the findings? Why is this important? Learn more at: https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2847514 Reference: Regev-Sadeh et al. JAMA Netw Open. 2026;9(4):e265648. PMID: 41949865. Through ICARE, we aspire to share cancer-related information …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post60226/
ICARE Social Media Post May 2026
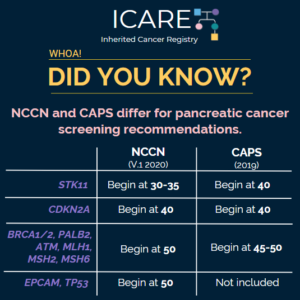
Low-Risk Pancreatic Cysts
A new study among over 6,000 patients with low-risk pancreatic cysts found that 0.6% developed pancreatic cancer, with a cancer risk rate 13x higher than the general population. What does this mean? Learn more at: https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2849164 Reference: Mirzaian et al. JAMA Netw Open. 2026;9(5):e2613808. PMID: 42160052. Through ICARE, we aspire to share cancer-related information that …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post052626/
ICARE Social Media Post May 2026
Breast Cancer Risk in Post-Menopausal Women
A new study showed that HIGHER body mass index (BMI) was linked to HIGHER risk for estrogen receptor positive (ER+) breast cancer. Learn more at: https://pubmed.ncbi.nlm.nih.gov/41650544/ Reference: Dauccia et al. Breast. 2026:104710. PMID: 41650544. Through ICARE, we aspire to share cancer-related information that may be of interest. If you have questions or clarifications, please direct …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post051926/
ICARE Social Media Post May 2026
Racial Disparities Across Subtypes in HR+ HER2- Breast Cancer
A recent study, led by our colleague Dr. Sonya Reid, among over 1,000 women with HR+ HER2- breast cancer found higher proportions of high-risk HR+ HER2- early-stage breast cancer among Black participants. Other findings include: These findings support the prognostic and potentially predictive importance of genomic testing to reduce racial survival disparities among Black women …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post051226/
ICARE Social Media Post May 2026
Ovarian Cancer Survivors
A new study shows that women with high grade serous ovarian cancer who remain cancer free for 7 years or more had nearly the same mortality risk as the general U.S. population. Of note, most of the women in this study had stage 3 ovarian cancer. Those with homologous repair deficiency (HRD) ovarian tumors (which …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post50526/
ICARE Social Media Post April 2026
ESMO Recommendations
The European Society of Medical Oncology (ESMO) evaluated breast cancer susceptibility genes on the basis of cancer-related mortality to help guide which genes should be included in genetic testing for inherited breast cancer. Those that may lead to patients living longer: BRCA1, BRCA2, PALB2, RAD51C, RAD51D, TP53 (for breast cancer diagnosed < age 40), and …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post42826/
ICARE Social Media Post April 2026
Exercise Lowers Risk of Dying from Cancer
New research suggests exercise (moderate to vigorous) LOWERS risk of dying from various types of cancers. Learn more at: https://pubmed.ncbi.nlm.nih.gov/41701497/ Reference: Rees-Punia et al. JAMA Netw Open. 2026;9(2):e2556971. PMID: 41701497. Through ICARE, we aspire to share cancer-related information that may be of interest. If you have questions or clarifications, please direct them to your healthcare …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post042226/
ICARE Social Media Post April 2026
Older Breast Cancer Survivors
A new study showed that chemotherapy through Taxanes (a common class of drugs used to treat breast cancer) was linked to higher risks for balance issues and neuropathy (but NOT to higher risk of falls) in women 65 and older at the time of breast cancer treatment. Learn more at: https://pubmed.ncbi.nlm.nih.gov/41719492/ Reference: Jackson et al. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post041426/
ICARE Newsletter Spring 2026
HOXB13 and Prostate Cancer
In a recently published study based on the VA’s Million Veteran Program, men with the HOXB13 p.G84E variant (the common variant in European populations, upon which much of the data is based) had a higher risk of prostate cancer, a slightly earlier age at diagnosis, and did not tend to develop more aggressive disease. Crawford, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nfs20266/
ICARE Newsletter Spring 2026
Introducing the NCCN Guidelines Navigator™
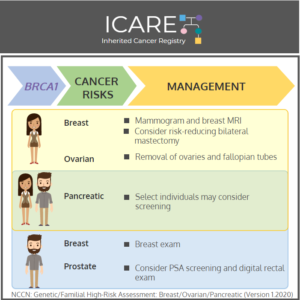
Explore this new interactive, color-coded platform designed to simplify navigation of NCCN’s Genetics/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate cancer guidelines. Whether you’re a clinician, researcher, or advocate, this tool helps you stay informed and make confident decisions in cancer care. To read more, create a FREE account at: https://www.nccn.org/guidelines/nccn-guidelines-navigator
Permanent link to this article: https://inheritedcancer.net/nls20262/
ICARE Newsletter Spring 2026
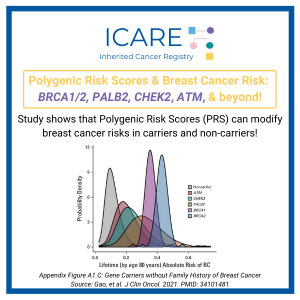
Consideration of Inherited Cancer Risk on a Continuum
Historically, heritable cancer risk was generally considered binary, based on the presence or absence of a germline pathogenic or likely pathogenic variant (GPV) in a known inherited cancer susceptibility gene (CSG). However, it has become clear that risk is more complex and presents on a continuum based on specific GPVs, in conjunction with interactions with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nfs20265/
ICARE Newsletter Spring 2026
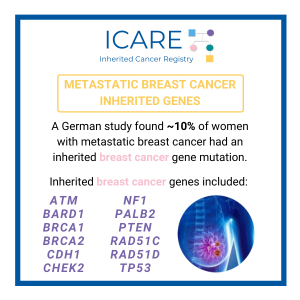
Genomics of Breast Cancers Among BRCA1/2, PALB2, CHEK2, and ATM Carriers
We recently published a study which included information from over 200 BRCA1, BRCA2, PALB2, CHEK2 and ATM carriers in ICARE who had breast cancer (mostly early-stage) combined with data from over 200 carriers (predominantly with metastatic breast cancer) who had clinical tumor testing. We pursued this research because we believe that distinct genomic changes in …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nfs202613/
ICARE Newsletter Spring 2026
New Gene Alert: SMARCAL1
A new study found the SMARCAL1 gene predisposes to a specific type of bone cancer (osteosarcoma) in children and young adults. Oak, et al. J Clin Oncol. 2025;43(36):3833-3843. PMID: 41066719. Article available at: https://pubmed.ncbi.nlm.nih.gov/41066719/. Social media post available at: https://www.facebook.com/share/v/1GZ2GdNU69/
Permanent link to this article: https://inheritedcancer.net/nfs20268/
ICARE Newsletter Spring 2026
BRCA1/2: Preventive Mastectomy vs. Screening
A new study shows similar rates of death from breast cancer when comparing 460 women who had a mastectomy with 745 women who chose surveillance (screening). For women choosing surveillance, these results are reassuring that survival is not likely to be compromised. Keep in mind, the goal of surveillance is to detect cancer early if …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nfs202610/
ICARE Newsletter Spring 2026
BRCA1/2: Preventive Bilateral Mastectomy Can Save Lives
A new study that combined the results of prior studies (through a systematic review/meta-analysis) including a total of over 6,000 BRCA1/2 carriers showed that preventive bilateral mastectomy lowered overall deaths as well as deaths from breast cancer, compared to not having this done. This information is important as women with BRCA1/2 mutations are deciding on …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nfs20269/
ICARE Newsletter Spring 2026
National Comprehensive Cancer Network (NCCN) Guideline Update Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, & Prostate
Select updates are outlined below. Check out the full guidelines by creating a FREE account at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bopp.pdf Version 2.2026 focused on adding comprehensive prostate cancer information and revising the name of the guidelines to encompass prostate cancer. Updates included: Version 3.2026 encompassed updates to the discussion section.
Permanent link to this article: https://inheritedcancer.net/nfs20263/
ICARE Newsletter Spring 2026
BRCA Carriers: High Risk for Ovarian Cancer After Breast Cancer Diagnosis
A new study of >2,000 BRCA1/2 carriers found 71 cases of ovarian/fallopian tube carcinoma following a breast cancer diagnosis. Of these 71 cases, 66 were among BRCA1 carriers and 5 were among BRCA2 carriers. BRCA1 carriers had a 5-fold higher risk of ovarian cancer compared to BRCA2 carriers. The risk of ovarian/fallopian tube cancer at …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nfs20264/
ICARE Newsletter Spring 2026
How Does Family History of Breast Cancers Affect Risk?
In a new study of almost 70,000 women, including those with hereditary forms of breast cancer, breast cancer risk was higher in those with a family history of breast cancer. Interestingly, family history raised breast cancer risks the least in BRCA1 carriers and the most in PALB2 carriers. Additionally, the effect of other risk factors …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nfs20267/
ICARE Newsletter Spring 2026
NCCN Guidelines Insights Article
JNCCN released an article in the February issue with insights on: Prostate cancer screening: Pancreatic cancer screening: Testing for non-epithelial ovarian cancer: Cheng, et al. J Natl Compr Cancer Netw. 2026:24(2):2-10. PMID: 41671423.Article available at: https://jnccn.org/view/journals/jnccn/24/2/article-p2.xml. Social media post available at: https://www.facebook.com/share/p/1Rk6tvn2AB/
Permanent link to this article: https://inheritedcancer.net/nls20261/
ICARE Newsletter Spring 2026
BRCA1/2: PARP Inhibitors
According to a new study, 50% of men in the U.S. with BRCA1/2 and metastatic castration-resistant prostate cancer receive PARP inhibitor treatment.1 This study also found that PARP inhibitor treatment differed by insurance coverage, with PARP inhibitors among commercially insured LESS LIKELY than those covered by government-issued insurance. Similarly, a U.S.-based study of adults with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nfs202612/
ICARE Newsletter Spring 2026
Ask the Expert
The following question was addressed by Joanne Kotsopoulos, PhD, who is a scientist at Women’s College Hospital in Toronto, Canada, with a longstanding interest in researching hormonal and other risk factors among those with inherited forms of breast cancer. Q:What is the latest information about risks of taking hormone replacement therapy (HRT) in BRCA1/2 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nfs202612-2/
ICARE Social Media Post March 2026
BRCA Carriers with Ovarian Cancer: Low Risk of Breast Cancer
A new study, which included ICARE participants, found that BRCA1/2 carriers with ovarian cancer have a relatively LOW risk of breast cancer. The hazard ratio for breast cancer after ovarian cancer compared to no ovarian cancer was 0.18 (95% CI: 0.12-0.27; P<0.0001). This means that getting breast cancer after already having ovarian cancer was MUCH …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post033126/
ICARE Social Media Post March 2026
BRCA1/2: Preventive Mastectomy vs. Screening
A new study shows similar rates of death from breast cancer when comparing 460 women who had a mastectomy with 745 women who chose surveillance (screening). Why is this important? Learn more at: https://ascopubs.org/doi/10.1200/JCO-25-00834 Reference: Gandhi, et al. J Clin Oncol. 2026:JCO2500834. PMID: 41637687. Through ICARE, we aspire to share cancer-related information that may be …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post032726/
ICARE Social Media Post March 2026
Somatic Alterations and Intrinsic Subtypes: Sporadic vs. Hereditary Breast Cancers
A new study among nearly 5,000 women with breast cancer, which included data from ICARE participants, compared sporadic cases to those with germline pathogenic or likely pathogenic variants in BRCA1, BRCA2, PALB2, ATM, and CHEK2. We studied these tumors because we hypothesized that breast cancers from BRCA1, BRCA2, PALB2, ATM, and CHEK2 carriers form by …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post032526/
ICARE Social Media Post March 2026
Inaccurate Recall of Inherited Cancer Genetic Test Results
A new study, which included ICARE participants, found that 11% of participants did not correctly recall their positive genetic test results. Those who did not correctly recall their result were more likely to have a history of cancer and not have a college. Inaccurate recall was also associated with: Why is this important?Knowing your genetic …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post032426/
ICARE Social Media Post March 2026
NCCN 2026 Breast Cancer Congress Presentation
Watch a valuable presentation from the NCCN 2026 Breast Cancer Congress by ICARE Principal Investigator and Clinical Geneticist, Dr. Tuya Pal, focused on breast cancer screening and genetic testing and how to optimize care for those at high risk 👇
Permanent link to this article: https://inheritedcancer.net/post031826/
ICARE Social Media Post March 2026
NCCN BOPP Guideline Update (v3.2026)
The National Comprehensive Cancer Network (NCCN) released an updated discussion section for the Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate Cancer guidelines on February 19th, 2026 (Version 3.2026), starting on Page 78 of the PDF. Check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_bopp.pdf Through ICARE, we aspire to share cancer-related information …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post031726/
ICARE Social Media Post March 2026
Take Charge of Your Cancer Risk: Webinar Recording
Watch the recording of a recent webinar, Take Charge of Your Cancer Risk: Learn What Your Genes Can Tell You, co-hosted by The Insighters and Susan G. Komen, during which ICARE Principal Investigator and Clinical Geneticist, Dr. Tuya Pal, shares insights on genetic testing for inherited cancer and strategies to reduce cancer risks 👇https://theinsighters.ewebinar.com/webinar/take-charge-of-your-cancer-risk-learn-what-your-genes-can-tell-you-22771/replay/22610921
Permanent link to this article: https://inheritedcancer.net/post031026/
ICARE Social Media Post March 2026
San Antonio Breast Cancer Symposium (SABCS) 2025 Recording
Don’t miss this summary of ICARE Founder Dr. Tuya Pal’s SABCS 2025 presentation, highlighting how genetic risk models can transform breast cancer prevention and care 👇https://tinyurl.com/BreastCancerRiskModels
Permanent link to this article: https://inheritedcancer.net/post30426/
ICARE Social Media Post Feburary 2026
Hysterectomy + Opportunistic Salpingectomy
A study of more than 80,000 women found an 80% lower risk of serous ovarian cancer (hazard ratio of 0.22) among those who had an opportunistic bilateral salpingectomy (i.e., removal of both fallopian tubes) when they were having a hysterectomy. Read the article to learn more: https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2844597 Reference: Sowamber, et al. JAMA Netw Open. 2026;9(2):e2557267. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post22426/
ICARE Social Media Post February 2026
NCCN Guidelines Insights Article
JNCCN just released an NCCN Guidelines insights article in the February issue with insights on prostate cancer screening, pancreatic cancer screening, and testing for non-epithelial ovarian cancer. Read it now at https://jnccn.org/view/journals/jnccn/24/2/article-p2.xml Reference: Cheng, et al. J Natl Compr Cancer Netw. 2026:24(2):2-10. PMID: 41671423.
Permanent link to this article: https://inheritedcancer.net/post21726/
ICARE Social Media Post February 2026
NCCN Guidelines Insights Article
JNCCN just released an NCCN Guidelines insights article in the February issue with insights on prostate cancer screening, pancreatic cancer screening, and testing for non-epithelial ovarian cancer. Read it now at https://jnccn.org/view/journals/jnccn/24/2/article-p2.xml Reference: Cheng, et al. J Natl Compr Cancer Netw. 2026:24(2):2-10. PMID: 41671423.
Permanent link to this article: https://inheritedcancer.net/post021726/
ICARE Social Media Post February 2026
Consideration of Inherited Cancer Risk on a Continuum
Preview our new article, which highlights key concepts in consideration of inherited cancer risk on a continuum at: https://www.gimjournal.org/article/S1098-3600(25)00306-5/fulltext Reference: Pal, et al. Genetics in Medicine, 2026, 101659, ISSN 1098-3600, https://doi.org/10.1016/j.gim.2025.101659.
Permanent link to this article: https://inheritedcancer.net/post20326/
ICARE Social Media Post January 2026
BRCA1/2: Preventive Bilateral Mastectomy Can Save Lives
A new study that combined the results of prior studies (through a systematic review/meta-analysis) including a total of over 6000 BRCA1/2 carriers showed that preventive bilateral mastectomy lowered overall deaths as well as deaths from breast cancer, compared to not having this done. This information is important as women with BRCA1/2 mutations are deciding on …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post012726/
ICARE Social Media Post January 2026
Breast Cancer Risk Stratification in Black Women
Check out our new article, which highlights the importance of early breast cancer risk assessment and the urgent need for inclusive research and better tools to improve health equity. Learn more at: https://doi.org/10.6004/jnccn.2025.7079 Reference: Reid S, Spalluto L, Pal T. Breast Cancer Risk Stratification in Black Women: Current Status and Potential Solutions to Improve Accuracy. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post01062026/
ICARE Social Media Post December 2025
Timing of BRCA Genetic Testing and Surgical Decision-Making
Check out our new article to answer the question… do Black women with breast cancer use genetic test results to make decisions about surgery? 👇 https://pmc.ncbi.nlm.nih.gov/articles/PMC12686358 Reference: Roberson, et al. Cancer Control. 2025:32:10732748251407739. PMID: 41360000.
Permanent link to this article: https://inheritedcancer.net/post12302025/
ICARE Social Media Post December 2025
BRCA1/2: HRT on Breast Cancer Risk
A new study among BRCA1/2 carriers without a prior breast cancer diagnosis, which included ICARE participants, showed that HRT use did NOT lead to a significant increase in breast cancer risk and estrogen alone HRT may be protective. Why is this important?Women with BRCA1/2 mutations have high risk of ovarian cancer; thus, removing the ovaries …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12232025/
ICARE Social Media Post December 2025
Alcohol and Cancer Risk
A new study showed that more than 50% of adults do NOT know that alcohol raises risk of cancer. In fact, findings showed that among those who drink alcohol, 76% thought there was no effect on cancer risk. Did you know that alcohol raises the risk of the following cancers? Learn more at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2840511 Reference: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12162025/
ICARE Social Media Post December 2025
New Gene Alert: SMARCAL1
A new study found the SMARCAL1 gene predisposes to specific type of bone cancer (osteosarcoma) in children and young adults. Learn more at: https://ascopubs.org/doi/10.1200/JCO-25-01114 Reference: Oak et al. J Clin Oncol. 2025. Online ahead of print. PMID: 41066719.
Permanent link to this article: https://inheritedcancer.net/post12092025/
ICARE Social Media Post December 2025
News Story: Woman’s Genetic Testing Discovery Highlights Importance of Family Health Conversations
ICARE participant, Sara Kavanaugh, shares her genetic testing journey with WSMV News 4 – a powerful reminder that family health conversations can save lives! Read the full article and watch the news segment, which also features ICARE founder Dr. Tuya Pal and ConnectMyVariant President Dr. Brian Shirts, at: https://www.wsmv.com/2025/11/18/nolensville-womans-genetic-testing-discovery-highlights-importance-family-health-conversations/
Permanent link to this article: https://inheritedcancer.net/post12032025/
ICARE Social Media Post December 2025
BRCA1/2: Metastatic Prostate Cancer
According to a new study, 50% of men in the U.S. with BRCA1/2 and metastatic castration-resistant prostate cancer receive PARP inhibitor treatment. This study also found that PARP inhibitor treatment differed by insurance coverage, with PARP inhibitors among commercially insured LESS LIKELY than those covered by government-issued insurance. Learn more at: https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2839543#xd_co_f=MTdiMGE0YjctNzA5ZC00YTFiLTkzMmQtNzBkZDIzZDg0NzEz~ Reference: Ostrowski et …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12022025/
ICARE Social Media Post December 2025
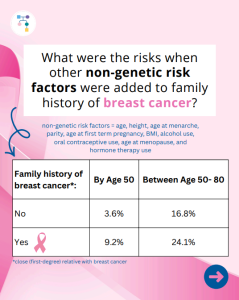
PALB2: How much does breast cancer family history and non-genetic risk factors affect risk?
A new study looked at breast cancer risks in PALB2 carriers based on family history and found that risks for breast cancer were much higher in those with a family history, while additional non-genetic risk factors seemed to minimally modify risks. To learn more read the article at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2839917 Reference: O’Brien, et al. JAMA Oncol. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12012025/
ICARE Social Media Post November 2025
HOXB13 and Prostate Cancer
In a recently published study based on the VA’s Million Veteran Program, men with the HOXB13 p.G84E variant had: Learn more at: https://jnccn.org/view/journals/jnccn/23/10/article-e257055.xml Reference: Crawford et al. J Natl Compr Canc Netw. 2025;23(10):e257055. PMID: 40953603.
Permanent link to this article: https://inheritedcancer.net/post11252025/
ICARE Social Media Post November 2024
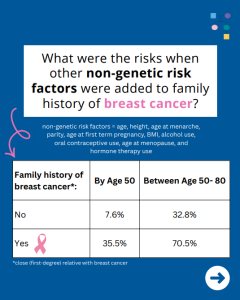
BRCA1: How much does breast cancer family history and non-genetic risk factors affect risk?
A new study reports that risks for breast cancer in BRCA1 carriers did NOT markedly differ based on family history. However, additional non-genetic risk factors were important modifiers of risk. To learn more read the article at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2839917 Reference: O’Brien, et al. JAMA Oncol. 2025. Online ahead of print. PMID: 41066089.
Permanent link to this article: https://inheritedcancer.net/post11242025/
ICARE Social Media Post November 2025
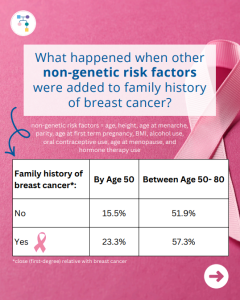
ATM: How much does breast cancer family history and non-genetic risk factors affect risk?
A new study evaluated breast cancer risks in ATM carriers based on family history and found that risks for breast cancer were higher in those with a family history and additional non-genetic risk factors modified risks in those who already had a family history. To learn more read the article at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2839917 Reference: O’Brien, et …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11212025/
ICARE Social Media Post November 2025
CHEK2: How much does breast cancer family history and non-genetic risk factors affect risk?
A new study looked at breast cancer risks in CHEK2 carriers based on family history and found that risks for breast cancer were much higher in those with a family history and additional non-genetic risk factors further modified risks. To learn more read the article at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2839917 Reference: O’Brien, et al. JAMA Oncol. 2025. Online …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11192025/
ICARE Social Media Post November 2025
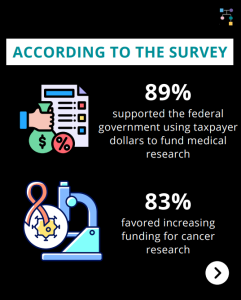
New AACR Poll Results
New poll results through the American Association for Cancer Research (AACR) showed that 9 in 10 Americans support federal funding for medical research. According to the survey: As a result of federally funded research, between July 2024 to June 2025, 32 new therapeutics, tests, and devices to treat or detect cancer were approved by the …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11182025/
ICARE Social Media Post November 2025
BARD1: How much does breast cancer family history and non-genetic risk factors affect risk?
A new study evaluated breast cancer risks in BARD1 carriers based on family history and found that risks for breast cancer were higher in those with a family history and additional non-genetic risk factors were important modifiers of risk. To learn more read the article at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2839917 Reference: O’Brien, et al. JAMA Oncol. 2025. Online …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11172025/
ICARE Social Media Post November 2025
BRCA2: How much does breast cancer family history and non-genetic risk factors affect risk?
A new study reports that risks for breast cancer in BRCA2 carriers were higher in those with a family history, while additional non-genetic risk factors seemed to minimally modify risks. To learn more read the article at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2839917 Reference: O’Brien, et al. JAMA Oncol. 2025. Online ahead of print. PMID: 41066089.
Permanent link to this article: https://inheritedcancer.net/post11142025/
ICARE Social Media Post November 2025
RAD51C: How much does breast cancer family history and non-genetic risk factors affect risk?
A new study evaluated breast cancer risks in RAD51C carriers based on family history and found that risks for breast cancer were higher in those with a family history, but only at later stages. Additional non-genetic risk factors were also important modifiers of risk. To learn more read the article at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2839917 Reference: O’Brien, et …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11122025/
ICARE Social Media Post November 2025
Weight loss drugs (GLP-1 Agonists) and Cancer Risks
Learn more by reading the full article at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2837870 Reference: Dai et al. JAMA Oncol. 2025:e252681. PMID: 40839273.
Permanent link to this article: https://inheritedcancer.net/post11112025/
ICARE Social Media Post November 2025
Li-Fraumeni Syndrome: Disclosure in New Romantic Partnerships
In a recent study among 33 individuals aged 15-39 years old with Li-Fraumeni syndrome, key factors in disclosure to new partners were: Disclosure was a process often done early to fulfill moral obligation and emotionally self-protect from future rejection. Why is this important? Learn more at https://pmc.ncbi.nlm.nih.gov/articles/PMC12423177/ Reference: Rising et al. Fam Cancer. 2025;24(4):71. PMID: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11042025/
ICARE Social Media Post October 2025
BRCA1/2, PALB2, CHEK2, & ATM: How does family history of breast cancers affect risk?
In a new study of almost 70,000 women, including those with hereditary forms of breast cancer, breast cancer risk was higher in those with a family history of breast cancer. Family history raised breast cancer risks: ✨Why is this important? ✨This type of information to personalize risk estimates can help women to guide screening and …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post10282025/
Permanent link to this article: https://inheritedcancer.net/post10262025/
ICARE Social Media Post October 2025
OCRA 2025 Presentation
Watch an exciting presentation by ICARE Principal Investigator and Clinical Geneticist, Dr. Tuya Pal, from the 2025 International Gynecologic Cancer Conference. During her talk, she highlights the critical role of genetic testing and ongoing follow-up care for individuals with hereditary cancer predisposition, and addresses the disparities that impact access to essential genetic services. 📺 Watch …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post10222025/
ICARE Social Media Post October 2025
Higher Rates of BRCA1/2 Mutations in Hispanic Women with Breast Cancer
New single institution study suggests HIGHER rates of BRCA1/2 mutations in Hispanic women with breast cancer. Specifically, a study of 2401 unselected breast cancer patients showed that Hispanic women were more than 2.5 times more likely to have a BRCA1/2 mutation compared to non-Hispanic women. BRCA1/2 mutations were found in: These findings show the importance …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post-10212025/
ICARE Social Media Post October 2025
Cuts for Federal Investments in Research and Development (Post #1)
What happens when federal investments in research and development are cut? Read the full report from the Information Technology & Innovation Foundation (ITIF) for more details:https://www2.itif.org/2025-reducing-federal-rd.pdf?et_rid=1130177171&et_cid=5740249
Permanent link to this article: https://inheritedcancer.net/post10202025/
ICARE Social Media Post October 2025
BRCA Carriers: High Risk for Ovarian Cancer After Breast Cancer Diagnosis
A new study of >2,000 BRCA1/2 carriers found 71 cases of ovarian/fallopian tube carcinoma following a breast cancer diagnosis. Of these 71 cases, 66 were among BRCA1 carriers and 5 were among BRCA2 carriers. BRCA1 carriers had a 5-fold higher risk of ovarian cancer compared to BRCA2 carriers. What was the overall risk in those …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post10142025/
ICARE Social Media Post October 2025
Real Pink Podcast: Decoding Genetic Testing
We’re proud to share an episode from the Susan G. Komen’s Real Pink podcast in which ICARE’s Principal Investigator, Dr. Tuya Pal, discusses the importance of genetic testing and early detection and decodes some common questions surrounding genetic testing. Listen to the full episode at ⤵https://tinyurl.com/realpink
Permanent link to this article: https://inheritedcancer.net/post10132025/
ICARE Social Media Post October 2025
NCCN Breast, Ovarian, Pancreatic, and Prostate Cancer Guidelines Update (V2.2026)
The National Comprehensive Cancer Network (NCCN) just released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate Cancer guidelines (Version 2.2026) today! Check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_bopp.pdf
Permanent link to this article: https://inheritedcancer.net/post10102025/
ICARE Social Media Post October 2025
NCCN Guideline Update: HOXB13
The National Comprehensive Cancer Network (NCCN) released updated prostate cancer guideline for HOXB13 in the new Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate Cancer guidelines (Version 2.2026) published today! Check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_bopp.pdf
Permanent link to this article: https://inheritedcancer.net/post10102025-2/
ICARE Social Media Post October 2025
ACMG Clinical Practice Resource on RAD51C, RAD51D, and BRIP1
Check out the recently released ACMG Clinical Practice Resource on RAD51C, RAD51D, and BRIP1, developed through a group of worldwide experts! 🌍👇 https://www.sciencedirect.com/science/article/pii/S1098360025002047
Permanent link to this article: https://inheritedcancer.net/post10092025/
ICARE Social Media Post October 2025
NCCN Navigator
🚀 Introducing the NCCN Guidelines Navigator™ – Version 1.2026! Explore this new interactive, color-coded platform designed to simplify navigation of NCCN’s Genetics/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate cancer guidelines. Whether you’re a clinician, researcher, or advocate, this tool helps you stay informed and make confident decisions in cancer care. Start exploring now by …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post10082025/
ICARE Social Media Post October 2025
Cancer Predisposition Genes: Risk Of Developing Single And Multiple Primary Cancers
Learn more by reading the full article at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2838068#xd_co_f=MTdiMGE0YjctNzA5ZC00YTFiLTkzMmQtNzBkZDIzZDg0NzEz~ Reference: Shevach et al. JAMA Oncol. 2025:e252879. PMID: 40875208.
Permanent link to this article: https://inheritedcancer.net/post10072025/
ICARE Social Media Post October 2025
NCCN CEG Guidelines (V1.2025): TP53 Post
The National Comprehensive Cancer Network (NCCN) just released updated Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Gastric Cancer guidelines on June 13th, 2025 (Version 1.2025). Included in these new guidelines are updates to the TP53 content as follows: To read more, check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_ceg.pdf
Permanent link to this article: https://inheritedcancer.net/post10062025/
ICARE Social Media Post October 2025
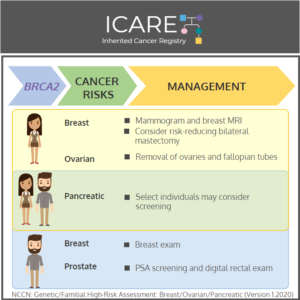
NCCN BOPP Guidelines (V1.2026): BRCA
In honor of Breast Cancer Awareness Month, we wanted to highlight the recently updated BRCA1/2 content in the National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate Cancer guidelines (Version 1.2026). To learn more, check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_bopp.pdf
Permanent link to this article: https://inheritedcancer.net/post10032025/
ICARE Newsletter Fall 2025
BRCA2 Gene: Mutation Type & Location Matter
A new study found that compared to those with BRCA2 mutations outside exon 11, those with exon 11 mutations had a lower breast cancer risk, higher risk for ER-negative breast cancer, and later age at diagnosis. These findings suggest that taking mutation type and location into account in cancer risk models may improve the ability …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nlf20255/
ICARE Newsletter Fall 2025
National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment Guideline Updates
Select updates outlined below. Check out the full guidelines by creating a FREE account at https://www.nccn.org/guidelines/category_2 Colorectal, Endometrial, & Gastric Cancer V1.2025 – Released June 13th, 2025 Breast, Ovarian, Pancreatic, & Prostate Cancer V1.2026 – Released July 10th, 2025
Permanent link to this article: https://inheritedcancer.net/nlf20251/
ICARE Newsletter Fall 2025
BRCA1/2 Carriers: High Risk of Breast Implant-Associated Lymphoma
In a new study of female BRCA1/2 carriers who had breast cancer, there was a 16-fold higher risk of anaplastic large-cell lymphoma associated with breast implants. This occurred in BRCA1/2 carriers who received textured breast implants as part of breast reconstruction. This is important information for BRCA1/2 carriers to know about to guide breast reconstruction …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nlf20254/
ICARE Newsletter Fall 2025
BRIP1: Third Most Common Gene for Inherited Ovarian Cancer
In a new study of unselected women with ovarian cancer, BRIP1 was the third most common gene for inherited ovarian cancer at a frequency of 1.1%, following BRCA1/2 which were found in 14.8%. Other inherited ovarian cancer genes included PALB2 (0.8%), RAD51C (0.4%), and RAD51D (0.4%). Beyond BRCA1/2 testing, additional testing through inherited cancer multi-gene …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nlf20256/
ICARE Newsletter Fall 2025
BRCA1/2 Carriers with Breast Cancer: Removal of Ovaries and Fallopian Tubes Lowers Risk of Death
A new study among BRCA1/2 carriers with breast cancer showed that the overall risk of death was 48% lower for those that removed their ovaries and fallopian tubes. Specifically, these women had lower risks of all-cause mortality, breast cancer-specific mortality, and second non-breast cancer development. Removing the ovaries leads to menopause which has other health …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nlf20252/
ICARE Newsletter Fall 2025
ATM, CHEK2, & PALB2 Carriers: Are There Differences in Cancer-Associated Mortality?
A new study of ATM, CHEK2, and PALB2 carriers compared to non-carriers showed similar mortality from breast cancer, pancreatic cancer, and colorectal cancer. Other findings among BRCA1/2 carriers and Lynch Syndrome patients showed: 1) BRCA1/2 carriers had lower mortality from triple-negative breast cancer; and 2) Lynch Syndrome patients had lower mortality from colorectal cancer. Ultimately, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nlf20257/
ICARE Newsletter Fall 2025
Ask the Expert
The question was addressed by Brian Shirts, MD, PhD, President of ConnectMyVariant and Service Medical Director of the Molecular Genetics Laboratories at Vanderbilt University Medical Center. If you have a question you would like addressed, email ICARE@vumc.org for consideration in future newsletters. Q: Why do you think cascade testing has not happened as much as …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nlf202511/
ICARE Newsletter Fall 2025
Ovarian Cancer: 1 in 4 Cases Could Have Been Prevented!
A new study of 1877 ovarian cancer patients showed almost 25% of patients had ‘missed opportunities’ for salpingectomy (removal of fallopian tubes) when they had another surgery or procedure before their ovarian cancer diagnosis. Additionally, 6% of patients had a close relative with ovarian cancer, and almost 20% had a mutation in an ovarian cancer …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nlf20253/
ICARE Newsletter Fall 2025
Lynch Syndrome: Showing the Importance of Family Testing
Cascade testing refers to testing family members for a gene mutation after another family member is found to have a mutation. Once family members get cascade testing, they can also benefit from screening, cancer prevention, and early detection strategies. A study conducted a microsimulation model to look at the cost effectiveness of cascade testing of …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nlf20258/
ICARE Newsletter Fall 2025
ICARE Community Spotlight: Kathy Baker
I was only 30 when my 32-year-old sister was diagnosed with breast cancer. A couple years later, when a mobile mammography bus showed up at my law school offering free mammograms, I decided it couldn’t hurt to be screened. When my mammogram was normal, I made plans to wait until age 50 for my next …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nlf202512/
ICARE Newsletter Fall 2025
Inherited Prostate Cancer: PARP Inhibitors
A new study combined the results of prior studies of PARP inhibitors in patients with metastatic castration-resistant prostate cancer and an inherited gene mutation through a meta-analysis. The results showed that PARP inhibitors provided the greatest benefit in BRCA1/2 carriers and there was a strong signal of benefit in PALB2 or CDK12 alterations. However, there …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nlf202510/
ICARE Newsletter Fall 2025
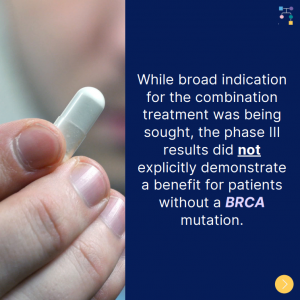
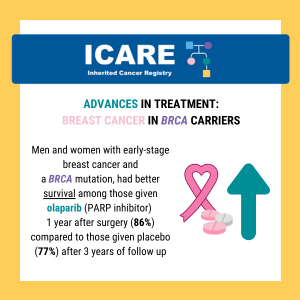
Breast Cancer Treatment: BRCA1/2 Carriers
A new randomized controlled trial among BRCA1/2 carriers comparing neoadjuvant chemotherapy with olaparib versus chemotherapy alone found: These findings suggest that adding olaparib may benefit survival for BRCA1/2 carriers, even if this is not apparent when looking at pathologic response. Abraham, et al. Nat Commun. 2025;16(1):4269. PMID: 40360463. Article available at: https://pubmed.ncbi.nlm.nih.gov/40360463/. Social media post …
Continue reading
Permanent link to this article: https://inheritedcancer.net/nlf20259/
ICARE Social Media Post September 2025
FDA Approval Update for Patients with Neurofibromatosis Type 1
Big news for the NF1 community: Selumetinib is now FDA-approved for inoperable plexiform neurofibromas. A major step forward in treatment options for Neurofibromatosis Type 1! 🔗 Read the full FDA announcement at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selumetinib-pediatric-patients-1-year-age-and-older-neurofibromatosis-type-1
Permanent link to this article: https://inheritedcancer.net/post09302025/
ICARE Social Media Post September 2025
Mammograms: Using AI to Distinguish DCIS from Invasive Breast Cancer
A new mammography study shows that artificial intelligence (AI) may enhance the differentiation of DCIS and invasive cancer from suspicious micro calcifications. In the future, this type of study might help better differentiate findings on mammograms.To learn more, read the full article at: https://www.academicradiology.org/article/S1076-6332(25)00732-9/abstractReference: Wenjie, X., et al. (2025). Differentiation of Suspicious Microcalcifications Using Deep …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post09232025/
ICARE Social Media Post September 2025
Ovarian Cancer: 1 in 4 cases could have been prevented!
Read the article to learn more! https://jamanetwork.com/journals/jamasurgery/article-abstract/2837473 Reference: Moufarrij, et al. JAMA Surg. 2025:e252810. PMID: 40802262.
Permanent link to this article: https://inheritedcancer.net/post09162025/
ICARE Social Media Post September 2025
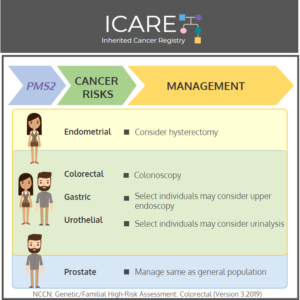
NCCN CEG Guidelines (V1.2025): PMS2 Post
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Gastric Cancer guidelines on June 13th, 2025 (Version 1.2025). Included in these new guidelines are updates to the surgical options for Lynch carriers and updates to the PMS2 content as outlined below: To read more, check out the full guidelines by creating …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post09152025/
ICARE Social Media Post September 2025
Bilateral Mastectomy Rates in Female Carriers with Breast Cancer
Learn more by reading the article at: https://link.springer.com/article/10.1245/s10434-025-17859-9 Reference: Kerivan, et al. Ann Surg Oncol. 2025. Online ahead of print. PMID: 40717162.
Permanent link to this article: https://inheritedcancer.net/post09092025/
ICARE Social Media Post September 2025
NCCN CEG Guidelines (V1.2025): EPCAM Post
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Gastric Cancer guidelines on June 13th, 2025 (Version 1.2025). Included in these new guidelines are updates to the surgical options for Lynch carriers and updates to the EPCAM content as outlined below: LS-C 1 of 5 (Page 50) LS-C 5 of 5 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post09062025/
ICARE Social Media Post September 2025
New MEN1 Guidelines
Learn more by reading the full article at: https://pubmed.ncbi.nlm.nih.gov/40232217/ Reference: Rivero, et al. Endocr Pract. 2025:S1530-891X(25)00038-2. PMID: 40232217.
Permanent link to this article: https://inheritedcancer.net/post09022025/
ICARE Social Media Post August 2025
NCCN CEG Guidelines (V1.2025): MSH2 Post
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Gastric Cancer guidelines on June 13th, 2025 (Version 1.2025). Included in these new guidelines are updates to the MSH2 content. To read more, check out the full guidelines by creating a FREE account at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_ceg.pdf
Permanent link to this article: https://inheritedcancer.net/post08272025/
ICARE Social Media Post August 2025
Risk Level: Risk-Reducing Mastectomy
Learn more by reading the full article at: https://jamanetwork.com/journals/jamaoncology/fullarticle/2836854
Permanent link to this article: https://inheritedcancer.net/post08262025/
ICARE Social Media Post August 2025
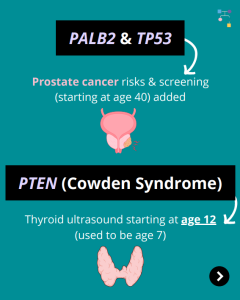
NCCN BOPP Guidelines (V1.2026): TP53
The National Comprehensive Cancer Network (NCCN) recently released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate Cancer guidelines (Version 1.2026), which included updates to TP53 content. To learn more, check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_bopp.pdf
Permanent link to this article: https://inheritedcancer.net/post082125/
ICARE Social Media Post August 2025
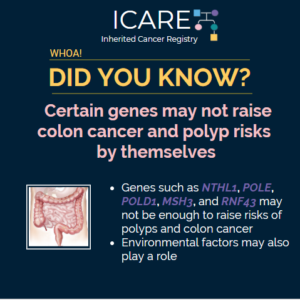
New Gene Alert: RPS20
🚨 New Gene Alert! 🚨RPS20 has been identified as a colorectal cancer risk gene: Learn more at:https://pubmed.ncbi.nlm.nih.gov/39920491/Reference: Amiot et al. Fam Cancer. 2025;24(1):22. PMID: 39920491. https://www.gimopen.org/article/S2949-7744(25)00981-1/fulltextReference: Herrera-Mullar et al. Genetics in Medicine Open. 2025;3(2):2949-7744. DOI: 10.1016/j.gimo.2025.102942
Permanent link to this article: https://inheritedcancer.net/post081925/
ICARE Social Media Post August 2025
NCCN BOPP Guidelines (V1.2026): PTEN
The National Comprehensive Cancer Network (NCCN) recently released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate Cancer guidelines (Version 1.2026), which included updates to PTEN content. To learn more, check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_bopp.pdf
Permanent link to this article: https://inheritedcancer.net/post081425/
ICARE Social Media Post August 2025
Lynch Syndrome: Low Dose Aspirin Lowers Colorectal Cancer Risk
According to new results of the Colorectal Adenoma/Carcinoma Prevention Program 3 (CaPP3) study presented at the International Cancer Prevention Conference in London, low-dose aspirin (100 mg) lowered the risk of colorectal cancer in patients with Lynch syndrome. Additionally, the use of aspirin lowered the risk of all cancers associated with Lynch syndrome. Learn more by …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post081225/
ICARE Social Media Post August 2025
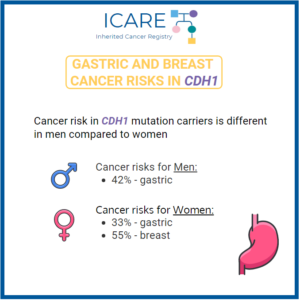
NCCN CEG Guidelines (V1.2025): CDH1 Post
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Gastric Cancer guidelines on June 13th, 2025 (Version 1.2025). Included in these new guidelines are updates to the CDH1 content as outlined below: HGAST-A (Page 97) HGAST-A (Page 97) To read more, check out the full guidelines by creating a FREE account …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post081125/
ICARE Social Media Post August 2025
NCCN BOPP Guidelines (V1.2026): CHEK2
The National Comprehensive Cancer Network (NCCN) recently released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate Cancer guidelines (Version 1.2026), which included updates to CHEK2 content. To learn more, check out the full guidelines by creating a FREE account at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bopp.pdf
Permanent link to this article: https://inheritedcancer.net/post080825/
ICARE Social Media Post August 2025
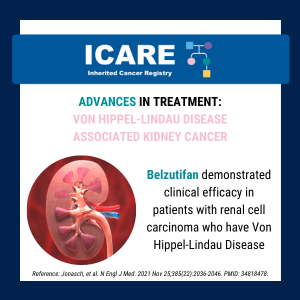
Von Hippel-Lindau (VHL)-related tumors and Belzutifan treatment
A new study presented at the American Society of Clinical Oncology (ASCO) annual meeting showed that Belzutifan treatment of VHL-related tumors durably shrinks tumors and may reduce the number of surgeries needed in patients with cancers associated with a von Hippel-Lindau (VHL) gene deletion or mutation. Five-year follow-up findings showed the following response rates: Learn …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post080525/
ICARE Social Media Post Month Year
NCCN CEG Guidelines (V1.2025): NTHL1 Post
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Gastric Cancer guidelines on June 13th, 2025 (Version 1.2025). Included in these new guidelines are updates to the NTHL1 content. To read more, check out the full guidelines by creating a FREE account at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_ceg.pdf
Permanent link to this article: https://inheritedcancer.net/post080425/
ICARE Social Media Post July 2025
BRCA1/2 Carriers: High Risk of Breast Implant-Associated Lymphoma
A new study among female BRCA1 and BRCA2 (BRCA1/2) carriers after breast cancer found that they had a 16-fold higher risk of anaplastic large-cell lymphoma associated with breast implants. This occurred in BRCA1/2 carriers who received textured breast implants as part of breast reconstruction. This is important information for BRCA1/2 carriers to know about to …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post072925/
ICARE Social Media Post July 2025
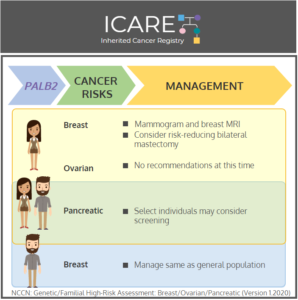
NCCN BOPP Guidelines (V1.2026): PALB2
The National Comprehensive Cancer Network (NCCN) recently released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate Cancer guidelines (Version 1.2026), which included updates to PALB2 content. To learn more, check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_bopp.pdf
Permanent link to this article: https://inheritedcancer.net/post072825/
ICARE Social Media Post July 2025
NCCN CEG Guidelines (V1.2025): STK11 Post
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Gastric Cancer guidelines on June 13th, 2025 (Version 1.2025). Included in these new guidelines are updates to the Peutz-Jeghers Syndrome (PJS) content. To learn more, check out the full guidelines by creating a FREE account at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_ceg.pdf
Permanent link to this article: https://inheritedcancer.net/post072525/
ICARE Social Media Post July 2025
BRCA1/2 Carriers with Breast Cancer: Removal of Ovaries and Fallopian Tubes Lowers Risk of Death
A new study among BRCA1/2 carriers with breast cancer showed that the overall risk of death was 48% lower for those that removed their ovaries and fallopian tubes. Specifically, these women had lower risks of: What about other health risks that can happen when ovaries are removed (causing menopause)? Why is this important? Learn more …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post072225/
ICARE Social Media Post Month Year
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update (V1.2025)
The National Comprehensive Cancer Network (NCCN) just released updated Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Gastric Cancer guidelines on June 13th, 2025 (Version 1.2025). There are many updates found in these new guidelines including updates to MSH2, EPCAM, PMS2, CDH1, NTHL1, and TP53 content as well as updates to the clinical diagnostic criteria for Peutz-Jeghers Syndrome. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post071425/
ICARE Social Media Post July 2025
NCCN Paper: Expansion of Recommendation for Hereditary Cancers
At the National Comprehensive Cancer Network (NCCN) 2025 Annual Conference, several experts in the field of inherited cancer discussed genes related to prostate, endometrial, and gastric cancers as well as how the NCCN Guidelines for Genetic/Familial High-Risk Assessment have been updated not only for healthcare providers, but also patients and their family members with similar …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post071225/
ICARE Social Media Post July 2025
NCCN Breast, Ovarian, Pancreatic, and Prostate Cancer Guidelines Update (V1.2026)
The National Comprehensive Cancer Network (NCCN) just released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate Cancer guidelines (Version 1.2026) today! There are many updates found in these new guidelines including updates to CHEK2, PALB2, TP53, and PTEN content as well as BRCA cancer risk management and much more. To learn more, check out …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post071025/
ICARE Social Media Post July 2025
NCCN Presentation: Expansion of Recommendation for Hereditary Cancers
Watch an exciting presentation from the National Comprehensive Cancer Network (NCCN) 2025 Annual Conference, during which several experts in the field of inherited cancer discuss genes related to prostate, endometrial, and gastric cancers as well as the expanded recommendations in the NCCN Guidelines for Genetic/Familial High-Risk Assessment. Watch now at ⤵https://education.nccn.org/node/97031
Permanent link to this article: https://inheritedcancer.net/post070925/
ICARE Social Media Post July 2025
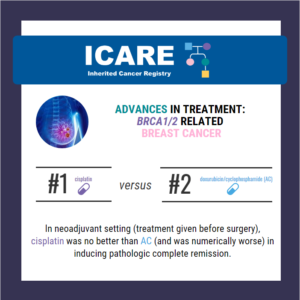
Breast Cancer Treatment: BRCA Carriers
A new randomized controlled trial among BRCA1 and BRCA2 carriers comparing neoadjuvant chemotherapy with olaparib versus chemotherapy alone found: This suggests that adding olaparib may benefit survival for BRCA carriers, even if this is not apparent when looking at pathologic response. Learn more by reading the full article at: https://pmc.ncbi.nlm.nih.gov/articles/PMC12075821/ Reference: Abraham, et al. Nat …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post070825/
ICARE Social Media Post June 2025
BRCA2 Gene: Mutation Type and Location Matter
A new study found that compared to those with BRCA2 variants outside exon 11, those with exon 11 mutations had: Why is this important? Learn more at: https://pubmed.ncbi.nlm.nih.gov/40288678/ Reference: Akamandisa, et al. Ann Oncol. 2025:S0923-7534(25)00170-X. Online ahead of print. PMID: 40288678.
Permanent link to this article: https://inheritedcancer.net/post062425/
ICARE Social Media Post June 2025
Polygenetic Risk Score (PRS) in Men with Prostate Cancer
A recent study showed that among men at highest risk for prostate cancer based on Polygenic Risk Score (PRS), more were found to have clinically significant disease (i.e., Gleason 7 or higher) than would have been found with PSA or MRI. In nearly 6,400 men who had PRS calculated:➡️ 745 (11.7%) had PRS in 90th …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post061725/
ICARE Social Media Post June 2025
Lynch Syndrome: Showing the Importance of Family Testing
Cascade testing refers to testing “at-risk” family members for a gene mutation, once the mutation has been found in a family member. For Lynch syndrome, once family members get cascade testing, they can also benefit from screening, cancer prevention, and early detection strategies. Findings of a microsimulation model looking at the cost effectiveness of cascade …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post061025/
ICARE Social Media Post June 2025
ATM, CHEK2, & PALB2 Carriers: Are There Differences in Cancer-associated Mortality?
A new study showed that compared to non-carriers, ATM, CHEK2, and PALB2 carriers showed similar mortality from breast cancer, pancreatic cancer, and colorectal cancer. Other findings among BRCA1/2 carriers and Lynch Syndrome patients showed: Why is this important?These results may be reassuring for ATM, CHEK2, and PALB2 carriers, and provide additional useful information when discussing …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post060625/
ICARE Social Media Post June 2025 2025
PREMM5: Model to Estimate the Risk for Having Lynch Syndrome
PREMM5 is a model to estimate the risk for having Lynch Syndrome. PREMMplus is a model that estimates risks in 19-cancer risk genes, including Lynch Syndrome genes, BRCA, and other genes. A new study that compared PREMM5 and PREMMplus found that PREMMplus was just as good as PREMM5 in identifying patients with Lynch Syndrome. PREMMplus …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post060325/
ICARE Social Media Post May 2025
Familial Adenomatous Polyposis (FAP): Risk-Reducing Colectomy
A new study among patients with Familial Adenomatous Polyposis (FAP), most of whom had a their colon removed to prevent cancer, found that although risk-reducing colectomy is now standard practice in FAP patients, colorectal cancer remains the most common cancer and cause of death in these patients. Recognizing cases of FAP that are ‘de novo’ …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post52725/
ICARE Social Media Post May 2025
BRIP1: Third Most Common Gene for Inherited Ovarian Cancer
A new study suggests that BRIP1 might be the THIRD most common gene for inherited ovarian cancer. BRCA1/2 mutations were found in almost 15%, while the frequency of mutations in other homologous recombination repair (HRR) genes among unselected ovarian cancer patients was: Why is this important?Testing in patients with ovarian cancer through inherited cancer multi-gene …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post52325/
ICARE Social Media Post May 2025
Inherited Prostate Cancer: PARP Inhibitors
A new meta-analysis study looking at prior studies of PARP inhibitors in patients with metastatic castration-resistant prostate cancer and an inherited gene mutation showed: Learn more at: https://pubmed.ncbi.nlm.nih.gov/39848867/ Reference: Naqvi, et al. Eur Urol. 2025:S0302-2838(24)02760-X. PMID: 39848867.
Permanent link to this article: https://inheritedcancer.net/post52125/
ICARE Social Media Post May 2025
Strategy to Intercept Cancers in Lynch Syndrome
Lynch syndrome related cancers have a distinct immune profile. They are generally “immunogenic”, meaning there might be opportunities to develop immune-interception strategies to prevent cancer. What is cancer “interception”? A new study based on work in mice showed that an EZH2 inhibitor (called GSK503) given over 9 weeks in mice lowered the number of adenomas …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post50525/
ICARE Social Media Post April 2025
Li-Fraumeni Syndrome: Decreased Time to Second Cancer in Children Treated with Radiation
A new case series of 47 children with Li-Fraumeni syndrome diagnosed with a solid cancer at or below age 16 found that: This study suggests that using radiation therapy to treat children with Li-Fraumeni syndrome and a solid tumor greatly raises the chance of getting a second cancer earlier. Therefore, it seems reasonable to only …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post42825/
ICARE Social Media Post April 2025
Colonoscopy Age in Lynch Syndrome: MSH6 and PMS2 carriers
A new study evaluating colonoscopy age in Lynch Syndrome patients with MSH6 and PMS2 mutations found that among MSH6 and PMS2 carriers, cancer was commonly found, even in those under age 30. Current recommendations suggest delaying colonoscopy until age 30 in these carriers, thus study authors suggest that this might lead to missed opportunities for …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post42725/
ICARE Newsletter Spring 2025
Community Spotlight
As someone with two inherited cancer gene mutations—MSH6 (Lynch syndrome) and CHEK2 — I know firsthand the emotional and practical complexities of navigating hereditary cancer risk. My journey began without what many might consider “classic” red flags — just a few scattered cancer cases in my family, none of which seemed connected at the time. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/12nls2025/
ICARE Newsletter Spring 2025
PARP Inhibitor: Relapsed BRCA-Mutated Ovarian Cancer
A new study (phase 3 ARIEL4 trial) to evaluate rucaparib (PARP inhibitor) versus standard-of-care chemotherapy among patients with relapsed BRCA-mutated ovarian cancer showed that median overall survival in the rucaparib group was 19.4 months versus 25.4 months in the chemotherapy group. This shows that more research is needed to figure out the most appropriate treatment …
Continue reading
Permanent link to this article: https://inheritedcancer.net/11nls2025/
ICARE Newsletter Spring 2025
New ASCO Guideline: Advanced Stage Ovarian, Fallopian Tube, and Peritoneal Cancer
A new guideline from the American Society of Clinical Oncology (ASCO) was released January 22nd, 2025, updating care for women with advanced-stage ovarian, fallopian tube, or primary peritoneal cancer. Recommendations included that these patients should be evaluated by a gynecologic oncologist before starting treatment to determine if they are candidates for primary cytoreductive surgery (meaning …
Continue reading
Permanent link to this article: https://inheritedcancer.net/10nls2025/
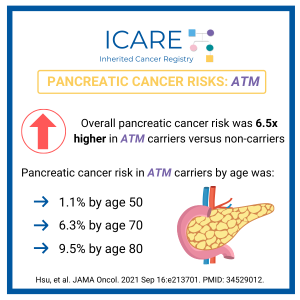
ICARE Newsletter Spring 2025
BRCA1/2: Pancreatic Cancer Risks in Women
A new international study in over 8000 BRCA1/2 carriers, which included data from ICARE participants, showed risk of pancreatic cancer to age 80 in BRCA1 was 2.2% (95% CI: 1.1%-4.3%) and in BRCA2 was 2.7% (95% CI: 1.3%-5.2%). Of the 34 BRCA1/2 carriers with pancreatic cancer, only 2 had a close (first-degree) relative with pancreatic …
Continue reading
Permanent link to this article: https://inheritedcancer.net/9nls2025/
ICARE Newsletter Spring 2025
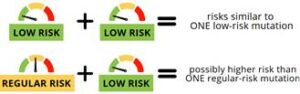
Refining Specific CHEK2 Mutation Risks
A recent editorial highlighted three common low-risk CHEK2 mutations (p.I157T, p.S428F, and p.T476M) that lead to a breast cancer risk of <1.4 fold (compared to “typical” CHEK2 mutations where the risk is over 2-fold).1 This is important because the level of risk for these mutations does not warrant high-risk screening. Another study on these three …
Continue reading
Permanent link to this article: https://inheritedcancer.net/8nls2025/
ICARE Newsletter Spring 2025
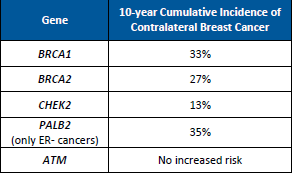
BRCA1/2: Second Primary Cancers After Breast Cancer
Through linking test results to electronic health records in England from 1995 to 2019, researchers estimated risks of a second primary cancer after breast cancer for BRCA1/2 carriers (Table 1) as well as risks over 10 years (Table 2). This study gives us more generalizable information about cancer risks to help guide risk assessment and …
Continue reading
Permanent link to this article: https://inheritedcancer.net/7nls2025/
ICARE Newsletter Spring 2025
Lynch Syndrome: Personalizing Risks
MyLynch is a resource for Lynch syndrome patients that provides personal cancer risks, education on interventions, and adjusted risk estimates, depending on the intervention(s) the patient chooses to pursue. If you have Lynch syndrome, go to https://hereditarycancer.dfci.harvard.edu/mylynch/ to get your personalized risk estimate. Check out a recent presentation by Dr. Yurgelun, who helped develop MyLynch, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nls2025/
ICARE Newsletter Spring 2025
Working Towards Defining a New Category of Reduced Penetrance BRCA1/2 Variants
We recently published a study that brings attention to “reduced penetrance” BRCA1 and BRCA2 (BRCA) mutations, which lead to LOWER breast cancer risks than “typical” BRCA mutations.1 Specifically, these mutations lead to a lifetime breast cancer risk of 20-30%, similar to moderate penetrance breast cancer genes such as CHEK2 or ATM. This level of risk …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nls2025/
ICARE Newsletter Spring 2025
ACMG Guidelines Focused on Risks and Care Among ATM Carriers
A panel of worldwide experts recently published recommendations for people with an ATM mutation, which raises the risk for breast, pancreatic, and prostate cancers. Among women Among men Among both women and men Check out an overview by Drs. Tischkowitz and Pal, who co-led this ATM effort, at: https://youtu.be/T76iGtn8_Do Pal T, et al. Genet Med. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nls2025/
ICARE Newsletter Spring 2025
New 2025 NCCN Guidelines for Patients
We are excited to announce the release of the newly created 2025 NCCN Guidelines for Patients: Genetic Testing for Hereditary Breast, Ovarian, Pancreatic, and Prostate Cancers. These comprehensive guidelines provide valuable insights and information for patients navigating genetic testing. You can view and download a free copy of the guidelines by visiting: www.nccn.org/patients/guidelines/content/PDF/genetics-patient.pdf
Permanent link to this article: https://inheritedcancer.net/3nls2025/
ICARE Newsletter Spring 2025
Therapeutic Clinical Trials for BRCA1 and BRCA2 Carriers
We are also excited to introduce a new initiative to help match BRCA1 and BRCA2 carriers to novel treatment trials for which they may be eligible. If you are interested, please scan the QR code or go to https://redcap.link/ICAREcontactform where you may also provide more details about your history and ask any specific questions you …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nls2025/
ICARE Newsletter Spring 2025
National Comprehensive Cancer Network (NCCN) Guideline Updates
Breast, Ovarian, Pancreatic, and Prostate Cancer Colorectal, Endometrial, and Gastric Cancer
Permanent link to this article: https://inheritedcancer.net/1nls2025/
ICARE Social Media Post April 2025
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update (V4.2024)
The National Comprehensive Cancer Network (NCCN) just released updated Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, Gastric Cancer guidelines on April 2nd, 2025 (Version 4.2024). In these updated guidelines, the discussion section has been updated starting on Page 74 of the PDF based on current guidelines. To read more, you can check out the full guidelines by …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post40725/
ICARE Social Media Post April 2025
MyLynch: Personalizing Lynch Syndrome Risks
Check out a resource called ‘MyLynch’, which provides people with Lynch syndrome with personal cancer risk estimates and ways to lower these risks! If you have Lynch syndrome, go to the following link to learn more about this resource and to get your personalized risk estimates:https://hereditarycancer.dfci.harvard.edu/mylynch/ You can also check out a recent presentation by …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post40425/
ICARE Social Media Post March 2025
PARP-Inhibitor: Relapsed BRCA-Mutated Ovarian Cancer
A new study (phase 3 ARIEL4 trial) to evaluate rucaparib (PARP Inhibitor) versus standard-of-care chemotherapy among patients with relapsed BRCA-mutated ovarian cancer showed median overall survival in the rucaparib group was 19.4 months versus 25.4 months in the chemotherapy group. This shows that more research is needed to figure out the most appropriate treatment options …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post32425/
ICARE Social Media Post March 2025
New Urine Test to Detect Prostate Cancer
A new study reported on an 18-gene ‘MyProstateScore 2.0’ Test which: This test measures 18 cancer-associated and high-grade cancer-associated genes in the urine. Learn more at ⬇️https://www.auajournals.org/doi/10.1097/JU.0000000000004421#xd_co_f=MTdiMGE0YjctNzA5ZC00YTFiLTkzMmQtNzBkZDIzZDg0NzEz~ Reference: Tosoian, et al. J Urol. 2025. PMID: 39836866.
Permanent link to this article: https://inheritedcancer.net/post31825/
ICARE Social Media Post March 2025
Assessing Molecular Subtypes of Young Black Females with Triple-Negative Breast Cancer
Our team’s newly published study among young Black women with triple-negative breast cancer (TNBC) found that: These findings underscore how TNBC subtypes may be used as prognostic biomarkers across populations. 📖 Read the full article here: https://www.nature.com/articles/s41523-025-00731-0 Reference: Rajagopal, et al. npj Breast Cancer 11, 28 (2025). https://doi.org/10.1038/s41523-025-00731-0
Permanent link to this article: https://inheritedcancer.net/post31325/
ICARE Social Media Post March 2025
Blood Test to Detect Early-Stage Pancreatic Cancer
A preliminary study reported that a blood test to detect early-stage pancreatic cancer achieved 98% specificity & 73% sensitivity! The test could accurately distinguish pancreatic ductal adenocarcinoma (PDAC) from other non-cancer pancreatic conditions. Learn more at ⬇️https://www.science.org/doi/10.1126/scitranslmed.adq3110 Reference: Montoya Mira, et al. Sci Transl Med. 2025;17(785):eadq3110. PMID: 39937880.
Permanent link to this article: https://inheritedcancer.net/post31125_1/
ICARE Social Media Post March 2025
NCCN BOPP Guideline Update (Version 3.2025)
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate Cancer guidelines on March 6th, 2025 (Version 3.2025), including updates to the discussion (starting on Page 74 of the PDF) based on the current guidelines that were updated over the past year. To read more, you can check out …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post31125/
ICARE Social Media Post February 2025
CHEK2: Double Mutation Carriers and Risks
There are three CHEK2 “low-risk” mutations with lower breast cancer risks: p.I157T, p.S428F, and p.T476M. A new study was conducted on how combinations of low- and regular-risk CHEK2 mutations may affect breast cancer risk. Results showed the following risks for various combinations of variants: Accompanying editorial by Dr. Rajagopal highlights: Check out the articles to …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post21025/
ICARE Social Media Post February 2025
ASCO Guideline Update: Advanced stage ovarian, fallopian tube or primary peritoneal cancer
The American Society of Clinical Oncology (ASCO) recently released a guideline update for advanced-stage ovarian, fallopian tube, or primary peritoneal cancer which recommends: These updates reinforce the importance of genetic testing and specialized evaluation in treatment planning, ensuring patients receive the most effective and personalized care. To view and download a copy for free, visit: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post20725/
ICARE Social Media Post February 2025
May 2025 BRCA Symposium
Calling all healthcare providers and researchers! 🧬 Join us at BRCA 2025 – the 10th International Symposium on Hereditary Breast and Ovarian Cancer, taking place from May 6th-9th, 2025 in Montréal, Québec, Canada. We’re celebrating 30 years since the discovery of the BRCA genes and 20 years of bringing the world’s top minds together to …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post20125/
ICARE Social Media Post January 2025
BRCA1/BRCA2 Carriers: Second Primary Cancers after Breast Cancer
Through linking test results to electronic health records in England from 1995 to 2019, researchers estimated relative and absolute second primary cancer risks for BRCA1/2 carriers after breast cancer. 📊 This study gives us more generalizable information about cancer risks in BRCA carriers, which can help guide risk assessment and management. Read the full article …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12825/
ICARE Social Media Post January 2025
Positive Gene Podcast
✨ New Episode! I had the privilege of visiting the Vanderbilt-Ingram Cancer Center in Nashville and sitting down with Dr. Tuya Pal, a leader in hereditary cancer genetics and the driving force behind ICARE Registry. We discuss the incredible work ICARE is doing to provide free resources, personalized updates, and opportunities to contribute to life-changing …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12325/
ICARE Social Media Post January 2025
NCCN Guidelines for Patients®: Genetic Testing for Hereditary Breast, Ovarian, Pancreatic, and Prostate Cancers
The National Comprehensive Cancer Network (NCCN) just posted the 2025 NCCN Guidelines for Patients: Genetic Testing for Hereditary Breast, Ovarian, Pancreatic, and Prostate Cancers. These patient guidelines are based on the NCCN Clinical Practice Guidelines in Oncology and explain expert recommendations for people with cancer and their caregivers! To view and download a copy for …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12225/
ICARE Social Media Post January 2025
Risk-reducing surgeries extend survival among young BRCA carriers with breast cancer history
🔬 A new study presented at the 2024 San Antonio Breast Cancer Symposium suggests that risk-reducing surgeries can improve survival for young BRCA carriers with a history of breast cancer. Among BRCA carriers with early breast cancer, risk-reducing surgeries (breast and/or ovaries) were found to: Read the full article to learn more at: https://www.healio.com/news/hematology-oncology/20241211/riskreducing-surgeries-extend-survival-among-young-brca-carriers-with-breast-cancer-history Reference: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12125/
ICARE Social Media Post January 2025
West African Genetic Ancestry and Breast Cancer Outcomes Among Black Women
Our team’s newly published study among young Black women with breast cancer found that: These findings underscore the need to go beyond self-reported race and consider genetic ancestry and social determinants of health in order to improve survival outcomes for Black women with breast cancer. 📖 Read the full article here: https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2827614 Reference: Reid, et …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11325/
ICARE Social Media Post January 2025
1 Year of Olaparib Improves Outcomes in BRCA Breast Cancer Patients
A new study presented at the 2024 San Antonio Breast Cancer Symposium suggests:⤷ Giving PARP Inhibitor (Olaparib) for one year after standard cancer treatment led to benefits in BRCA-mutation breast cancers. What was seen? These findings bring up the possibility of considering using PARP inhibitors in BRCA carriers: To learn more, read the full article …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post10925/
ICARE Social Media Post January 2025
VUS Pilot Study Recruitment Post
📣 Are you or someone you know living with a Variant of Uncertain Significance (VUS) in an inherited cancer gene? We’re inviting you to enroll into ICARE to be considered for a valuable research effort focused on improving resources for those with a VUS result. When you participate in ICARE, you’ll:✔️ Be considered for this …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post10425/
ICARE Social Media Post January 2025
May 2025 BRCA Symposium
Calling all healthcare providers and researchers! 🧬 Join us at BRCA 2025 – the 10th International Symposium on Hereditary Breast and Ovarian Cancer, taking place from May 6th-9th, 2025 in Montréal, Québec, Canada. We’re celebrating 30 years since the discovery of the BRCA genes and 20 years of bringing the world’s top minds together to …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post10125/
ICARE Social Media Post December 2024
RAD51C VUS Results
🔬A new study uses additional laboratory analyses to figure out which Variant of Uncertain Significance (VUS) results may actually be mutations (i.e., positive results): 📈 Why is this important? Read the full article to learn more at: https://www.sciencedirect.com/science/article/pii/S0092867424009681 Reference: Olvera-León, et al. Cell. 2024;187(20):5719-5734.e19. PMID: 39299233.
Permanent link to this article: https://inheritedcancer.net/post122324/
ICARE Social Media Post December 2024
BRCA Carriers: Risks with the pill (Hormonal Contraception)
A recent study found that among women with BRCA1 and BRCA2 mutations, oral contraceptive use:🧬 Increased risk of breast cancer with proportionate increases of 3% for each year of hormonal contraceptive use among BRCA1 carriers.🧬 No association seen for BRCA2 carriers. 📈 Why is this important? Read the full article to learn more at: https://pubmed.ncbi.nlm.nih.gov/39356978/ …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post121824/
ICARE Social Media Post December 2024
Pancreatic Cancer in Women with BRCA1 and BRCA2
A new international study in over 8000 BRCA1/2 carriers, including ICARE participants, showed lifetime pancreatic cancer risk to age 80 in: 👩👩👧👦 Of the 34 BRCA carriers with pancreatic cancer, 2 reported a first-degree relative with pancreatic cancer. ⚠️ Risk factors for pancreatic cancer include alcohol intake and history of diabetes. 📈 5-year survival rate …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post121224/
ICARE Social Media Post December 2024
ACMG Clinical Practice Resource on ATM
Check out the recently released ACMG Clinical Practice Resource on ATM, developed through a group of worldwide experts! 🌍👇 https://authors.elsevier.com/c/1kD7w3vlFV4Eph
Permanent link to this article: https://inheritedcancer.net/post120524/
ICARE Social Media Post December 2024
Alu-Mediated Breakpoints in VHL Pathogenic Variants
A study on mutations in Von-Hippel-Lindau (VHL) syndrome families revealed that: What does this mean?• In families that meet clinical criteria for VHL without a detectable VHL pathogenic variant whole genome sequencing may be considered for further analysis To learn more, read the full article at:https://pubmed.ncbi.nlm.nih.gov/33675279/ Reference: Vocke CD, et al. Hum Mutat. 2021; 42(5):520-529. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post120424/
ICARE Social Media Post December 2024
VHL Syndrome Characteristics
A recent study in carriers with Von-Hippel-Lindau Syndrome, an inherited condition that causes tumors and cysts to grow in certain areas of the body, found:▪️ Nearly 100% of carriers show some signs of this condition by age 65▪️ Demonstrates that this condition is highly penetrant Read the full article to learn more at:https://pubmed.ncbi.nlm.nih.gov/34916234/ Reference: Zhang …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-social-media-post-december-2024-vhl-syndrome-characteristics/
ICARE Social Media Post November 2024
BRCA1/2 Carriers with Advanced Breast Cancer
As highlighted in our latest ICARE newsletter, a new study in BRCA1/2 carriers with advanced breast cancer found: Read the full article to learn more at: https://pubmed.ncbi.nlm.nih.gov/37437366/Reference: Valenza, et al. Eur J Cancer. 2023;190:112944. PMID: 37437366. We also encourage you to read the full ICARE newsletter for other clinical and research updates at https://inheritedcancer.net/newsletters/
Permanent link to this article: https://inheritedcancer.net/icare-social-media-post-november-2024-brca1-2-carriers-with-advanced-breast-cancer/
ICARE Social Media Post November 2024
Breast Cancer After Ovarian Cancer in BRCA Carriers
As highlighted in the latest ICARE newsletter, a recent study evaluated breast cancer risks after ovarian cancer in BRCA1 & BRCA2 carriers. 🔍 After chemotherapy for ovarian cancer: 📊 Incidence rates were: 📈 What does this mean? Learn more at: https://tinyurl.com/yc8de3py Reference: Evans, et al. Genet Med. 2024;26(9):101172. PMID: 38847192. We also encourage you to …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post111924/
ICARE Social Media Post November 2024
Germline Mutations in Renal Cell Cancer Patients
A new study among patients with renal cell carcinoma reported a higher chance of finding a mutation in an inherited cancer-related gene in those with: Read the full article to learn more at:https://pubmed.ncbi.nlm.nih.gov/38127826/ Reference: Nguyen, et al. JCO Precis Oncol. 2023;7: e2300168. PMID: 38127826.
Permanent link to this article: https://inheritedcancer.net/post111424/
ICARE Social Media Post November 2024
BRCA Carriers with Breast Cancer: Trial to Compare PARP Inhibitors to Chemotherapy
As highlighted in the latest ICARE newsletter, a recent trial among BRCA1/2 carriers with breast cancer comparing PARP inhibitors to standard chemotherapy (Treatment of Physician’s Choice – TPC) found that after 25.7 months of follow up: Overall survival in each group was: The % alive at 3 years was: Patients who received Olaparib for first …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post111224/
ICARE Social Media Post November 2024
Reduced Penetrance BRCA1/2 Pathogenic Variants
Check out our recently published study that brings attention to “reduced penetrance” BRCA1 and BRCA2 (BRCA) pathogenic variants, which impart LOWER breast cancer risks than ‘typical’ BRCA mutations. Specifically, lifetime breast cancer risks for these reduced penetrance BRCA variants are 20-30% which is similar to that seen in moderate penetrance breast cancer genes (e.g., CHEK2 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post111124/
ICARE Social Media Post November 2024
NCCN Breast, Ovarian, Pancreatic, and Prostate Cancer Guidelines Update
The National Comprehensive Cancer Network (NCCN) just released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic, and Prostate Cancer guidelines on November 7th, 2024 (Version 2.2025) You read that right – they have now added “prostate” to the name as well as the relevant content! Prostate cancer specific testing and risk management information is now centralized …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post110824/
ICARE Social Media Post November 2024
NCCN Guideline Updates: CDKN2A
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer guidelines on September 11th, 2024 (Version 1.2025), which included the following CDKN2A updates ⤸ The CDKN2A gene produces two isoforms, both of which are tumor suppressors:➡️ p16INK4A and p14ARF For individuals with both of these isoforms, recommend:➡️ Skin exams …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post110724/
ICARE Social Media Post November 2024
NCCN BOP Guideline Update #10: BRCA2 Update
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer guidelines on September 11th, 2024. In these updates, NCCN added consideration of pancreatic cancer screening for BRCA2 carriers as outlined in PANC-A 1 of 2 (Page 52). To read more, you can check out the full guidelines by creating …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post110224/
ICARE Social Media Post November 2024
May 2025 BRCA Symposium
Calling all healthcare providers and researchers! 🧬 Join us at BRCA 2025 – the 10th International Symposium on Hereditary Breast and Ovarian Cancer, taking place from May 6th-9th, 2025 in Montréal, Québec, Canada. We’re celebrating 30 years since the discovery of the BRCA genes and 20 years of bringing the world’s top minds together to …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post110124/
ICARE Social Media Post October 2024
Personalized Screening Recommendations for CHEK2 Carriers
A recent editorial highlights the value of personalized screening recommendations for CHEK2 carriers▪️ Notably, three common low-risk mutations (p.I157T, p.S428F, and p.T476M) impart a breast cancer risk of less than 1.4 fold▪️ This level of risk does not warrant high-risk breast screening or other heightened care Read the full article to learn more at: https://www.oncotarget.com/article/28604/pdf/ …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post103124/
ICARE Social Media Post October 2024
Men’s Stories About Hereditary Cancer
In recognition of Breast Cancer Awareness Month, we are excited to share the newly released “Men’s Stories About Hereditary Cancer”. Read the full piece to hear how BRCA gene mutations affect men and steps you can take to learn about and manage your cancer risks ⤸https://tinyurl.com/mensstorieshereditarycancer
Permanent link to this article: https://inheritedcancer.net/post102824/
ICARE Social Media Post October 2024
NCCN BOP Guideline Update #9: Pancreatic Cancer Screening Update
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer guidelines on September 11th, 2024. An important update includes consideration of pancreatic cancer screening for ATM and BRCA2 carriers even in the absence of family history as outlined in PANC-A 1 of 2 (PAGE 52). To read more, you …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102824_1/
ICARE Social Media Post October 2024
NCCN BOP Guideline Update #8: CHEK2 Bi-allelic Mutations
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer guidelines on September 11th, 2024. In these updates, NCCN outlines emerging data regarding breast cancer risks for bi-allelic CHEK2 mutations (i.e., mutations in both copies of the gene) as outlined in GENE-A (Page 38) ⤸ To read more, you …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102424/
ICARE Social Media Post October 2024
Lack of Genetic Counseling and Testing Among Female Cancer Survivors
A recent study among women with early-stage breast cancer revealed that many female cancer survivors who meet guidelines for genetic counseling and testing for inherited cancer do not receive it. These findings underscore the need for better access to genetic counseling and testing to support patients and their families. Learn more at: https://bit.ly/3MkeM2o Reference: Katz, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102324/
ICARE Social Media Post October 2024
NCCN BOP Guideline Update #7: CHEK2: Ile157Thr and Ser428Phe
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer guidelines on September 11th, 2024. In these updates, NCCN outlines the following for the CHEK2 Ile157Thr and Ser428Phe variants as outlined in GENE-A (Page 38) ⤸ To read more, you can check out the full guidelines by creating a …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102324_1/
ICARE Social Media Post October 2024
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update V2.2024 – #2 EPCAM Updates
The National Comprehensive Cancer Network (NCCN) released updated Genetic Familial High-Risk Assessment Colorectal, Endometrial, and Gastric Cancer guidelines on October 3rd, 2024. In these updated guidelines, NCCN revised information about EPCAM gene (which has usually been lumped together with MSH2) as follows ⤸ You can check out the full guidelines by creating a FREE account …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102224/
ICARE Social Media Post October 2024
Inherited Breast Cancers in Men
In honor of Breast Cancer Awareness month, we wanted to spread awareness about inherited breast cancer risks among men. Did you know that men with a BRCA2 mutation have about a 7% lifetime risk of breast cancer and those with a BRCA1 or PALB2 mutation have about a 1% lifetime risk of breast cancer? Thus, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102124/
ICARE Social Media Post October 2024
NCCN BOP Guideline Update #6: BRCA1 R1699Q
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer guidelines on September 11th, 2024. Some BRCA1 updates as outlined in GENE-A (Page 36) include ⤸ To read more, you can check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf
Permanent link to this article: https://inheritedcancer.net/post101924/
ICARE Social Media Post October 2024
November 2024 BRCA Hybrid Event Post – Patients & Public (Nov 5th) First post
Are you or someone you love affected by a BRCA gene mutation 🧬 ? This event is for you! On November 5th, Women’s College Hospital will be hosting a hybrid event entitled, BRCA: 30 Years, Discovery to Impact, to celebrate 30 years since the groundbreaking discovery of the BRCA1 and BRCA2 genes. Learn from leading …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post101724/
ICARE Social Media Post October 2024
NCCN BOP Guideline Update #5: ATM Update
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer guidelines on September 11th, 2024. In these new guidelines, there were several updates for ATM carriers, including adding colorectal cancer risk as outlined in GENE-A (Page 35) and consideration of pancreatic cancer screening as outlined in PANC-A 1 of …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post101724_1/
ICARE Social Media Post October 2024
NCCN BOP Guideline Update #4: Ovarian Cancer Risks and Management Updates
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer guidelines on September 11th, 2024. Some updates include revisions to ovarian cancer risks and management including ⤸ To read more, you can check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf
Permanent link to this article: https://inheritedcancer.net/post101224/
ICARE Social Media Post October 2024
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update V2.2024 = #1 Testing Considerations
The National Comprehensive Cancer Network (NCCN) released updated Genetic Familial High-Risk Assessment Colorectal, Endometrial, and Gastric Cancer guidelines on October 3rd, 2024. In these updated guidelines, NCCN added the following to testing being considered ⤸Personal history of colorectal or endometrial cancer at or older than age 50, and: You can check out the full guidelines …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post101524/
ICARE Social Media Post October 2024
Pancreatic Cancer Screening in High-Risk Patients
A recent study among high-risk patients found that pancreatic cancer screening through endoscopic ultrasound and/or MRI-based tests led to detection of smaller and earlier-stage cancers and overall, seemed to result in longer survival. Patients with an inherited gene mutation that predisposes to pancreatic cancer and/or a family history of pancreatic cancer were considered to be …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post101024/
ICARE Social Media Post October 2024
Double Mastectomy Not Linked To Survival Advantage
A recent study found that in women with breast cancer, double mastectomy did NOT lead to longer survival. This is very important information for women to know as they make decisions about breast cancer surgery. Read the full article to learn more at: https://bit.ly/47a5O1b Reference: Giannakeas, et al. JAMA Oncol. 2024:e242212. PMID: 39052262.
Permanent link to this article: https://inheritedcancer.net/post100924_1/
ICARE Social Media Post October 2024
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update V2.2024
The National Comprehensive Cancer Network (NCCN) released updated Genetic Familial High-Risk Assessment Colorectal, Endometrial, and Gastric Cancer guidelines on October 3rd, 2024. Updates include ⤸ Added the following to testing being considered: Personal history of colorectal or endometrial cancer at or older than age 50, and: Revised information about EPCAM gene (which has usually been …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post100824/
ICARE Social Media Post October 2024
NCCN BOP Guideline Update #3: Genetic Testing in Breast Cancer Patients Age 65 and Below
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer guidelines on September 11th, 2024. In these updated guidelines, NCCN indicates testing criteria for high-penetrance breast cancer susceptibility genes should be considered for those with a personal history of breast cancer ≤ age 65 (used to be < age …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post100824_1/
ICARE Social Media Post October 2024
BRCA2: Male Breast Cancer Risks
Did you know that men can get breast cancer too? In fact, Beyonce’s father, Matthew Knowles, was diagnosed with breast cancer in October 2019 and was found to have a BRCA2 mutation. Matthew’s children have a 50/50 chance of inheriting his BRCA2 mutation. Men with an inherited BRCA2 mutation are at an increased risk for …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post100724/
ICARE Social Media Post October 2024
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update – VERSION 5
The National Comprehensive Cancer Network (NCCN) released the new Genetic/Familial High-Risk Assessment Colorectal, Endometrial, and Gastric Cancer guidelines on August 8th, 2024. An important update includes the revision of CHEK2 estimated absolute colon cancer risk to “NO INCREASED RISK”; thus, general population screening is appropriate for these individuals. You can check out the full guidelines …
Continue reading
Permanent link to this article: https://inheritedcancer.net/100624-2/
ICARE Social Media Post October 2024
NCCN BOP Guideline Update #2: Testing Unaffected Family Members
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer guidelines on September 11th, 2024. In these updates, NCCN clarified that it is appropriate to test unaffected (not best testable) family members when they meet testing criteria. To read more, you can check out the full guidelines by creating …
Continue reading
Permanent link to this article: https://inheritedcancer.net/100424_1/
ICARE Social Media Post October 2024
NCCN BOP Guideline Update #1: Point-of-Care Testing
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer guidelines on September 11th, 2024. In these updated guidelines, the minimal components that should be discussed during point-of-care testing are outlined (found in EVAL-A 10 of 11; Page 17). To read more, you can check out the full guidelines …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post100324/
ICARE Newsletter Fall 2024
Community Spotlight: The Patient and the Researcher Shares Her Uncertain Future and Lessons She’s Learned
By Marleah Dean Kruzel, PhD, BRCA2 Previvor When I was eight years old, my mother found a lump in her breast – barely noticeable. For a few years, I watched her undergo chemotherapy, radiation, and a prophylactic mastectomy and reconstruction. Since then, my maternal aunt and grandmother also fought breast cancer, and we learned my …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2024-community-spotlight-the-patient-and-the-researcher-shares-her-uncertain-future-and-lessons-shes-learned/
ICARE Newsletter Fall 2024
Double Mastectomy Not Linked to Survival Advantage
A recent study found that in females with unilateral breast cancer (meaning breast cancer on one side) who decided to have a double mastectomy had similar mortality rates to those treated with lumpectomy or unilateral mastectomy (meaning double mastectomy did NOT lead to longer survival). Findings showed that while bilateral mastectomy greatly lowered the risk …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2024-double-mastectomy-not-linked-to-survival-advantage/
ICARE Newsletter Fall 2024
New Study to Evaluate Pancreatic Cancer Screening
A recent study among high-risk patients found that pancreatic cancer screening through endoscopic ultrasound and/or MRI-based tests led to detection of smaller and earlier-stage cancers, and overall, seemed to result in longer survival. Patients with an inherited gene mutation that predisposes to pancreatic cancer and/or a family history of pancreatic cancer were considered to be …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2024-new-study-to-evaluate-pancreatic-cancer-screening/
ICARE Newsletter Fall 2024
BRCA1/2 Carriers: Treatment Advances
Among BRCA1/2 carriers with advanced breast cancer, PARP inhibitors showed some activity even in patients with platinum resistant/unresponsive disease. However, the optimal delivery of platinum agents and PARP inhibitors was not clear.1 In another study of BRCA1/2 carriers with breast cancer (OlympiAD Trial), PARP inhibitors were compared to chemotherapy Treatment of Physician’s Choice (TPC), with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2024-brca1-2-carriers-treatment-advances/
ICARE Newsletter Fall 2024
American Society of Clinical Oncology (ASCO) Guideline Update: Selecting Genetic Tests in Patients with Various Cancers
What elements are most important in the collection of family history? When and how to use multigene panel testing when indicated? Which genes should be tested based on cancer type? Among patients with tumor testing, who should be offered germline testing? For a full list of recommendations in this guideline, the article is available at: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2024-american-society-of-clinical-oncology-asco-guideline-update-selecting-genetic-tests-in-patients-with-various-cancers/
ICARE Newsletter Fall 2024
Breast Cancer After Ovarian Cancer in BRCA1/2 Carriers
A recently published study reported that among females with ovarian cancer who received chemotherapy, their risk for breast cancer was lower for the next 5 years. Specifically, incidence rates were lower at 2 years (1.18%) and between 2 to 5 years (1.13%); however, incidence rates rose thereafter for BRCA1 carriers (>4% annually post 10 years). This study …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2024-breast-cancer-after-ovarian-cancer-in-brca1-2-carriers/
ICARE Newsletter Fall 2024
BRCA1/2 Carriers and Pregnancy-Related Risk
A recent study reported that pregnancy after breast cancer was safe for both mother and baby.1 Specifically, the researchers found that pregnancy after breast cancer was not associated with adverse maternal prognosis or fetal outcomes. Another study reported that breast cancer after pregnancy could be associated with poorer outcomes. Specifically, breast cancer diagnosed within 10 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2024-brca1-2-carriers-and-pregnancy-related-ris/
ICARE Newsletter Fall 2024
BRCA1/2 Carriers: Risk of Peritoneal Cancer After Bilateral Oophorectomy
A recent study among BRCA1 and BRCA2 carriers, which included ICARE participants, found that the risk for peritoneal cancer following a preventive bilateral oophorectomy was higher among BRCA1 carriers than BRCA2 carriers. Specifically, among 6310 females, the annual risk of peritoneal cancer was 0.14% for BRCA1 carriers and 0.06% for BRCA2 carriers, and the 20-year …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2024-brca1-2-carriers-risk-of-peritoneal-cancer-after-bilateral-oophorectomy/
ICARE Newsletter Fall 2024
Risk of Melanoma in BRCA1/2 Carriers
Over the last few years, there has been mixed information about the risk of melanoma in BRCA1/2 carriers – while earlier studies had suggested an association,1,2 other studies showed no association.3,4 Thus, based on current evidence, it is unclear if BRCA1 and/or BRCA2 pathogenic variants increase melanoma risk based on the current evidence.5,6 However, more recently, a study, which …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2024-risk-of-melanoma-in-brca1-2-carriers/
ICARE Newsletter Fall 2024
BRCA2-Associated Prostate Cancer
A new study found that males with a BRCA2 mutation and metastatic hormone-sensitive prostate cancer face poorer outcomes. BRCA2 carriers were also found to have a higher risk of progression to castration-resistant prostate cancer. These findings highlight the importance of evaluating germline mutations among prostate cancer patients. Custodio-Cabello, et al. Urol Oncol. 2024;42(10):331.e13-331.e24. PMID: 38926076. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2024-brca2-associated-prostate-cancer/
ICARE Newsletter Fall 2024
Risks of Cancer in Individuals with TWO CHEK2 Mutations (called “Bi-Allelic” Mutations)
A new study found that risks may be higher for multiple types of cancer, including breast cancer, in both females and males who have two CHEK2 mutations: Additionally, findings showed that the type of mutation may be important to guide level of risk (e.g., truncating 1100delC homozygotes being at a higher risk than compound heterozygotes …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2024-risks-of-cancer-in-individuals-with-two-chek2-mutations-called-bi-allelic-mutations/
ICARE Newsletter Fall 2024
National Comprehensive Cancer Network (NCCN) Guideline Updates
Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer – Released September 11th, 2024 (V1.2025) Check out the full guidelines by creating a FREE account at www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf Testing Updates: Gene Updates: Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, and Gastric Cancer – Released August 8th, 2024 (V1.2024) Check out the full guidelines by creating a FREE account at …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2024-national-comprehensive-cancer-network-nccn-guideline-updates/
ICARE Social Media Post September 2024
BRCA2-Associated Prostate Cancer
A new study found that men with a BRCA2 mutation and metastatic hormone-sensitive prostate cancer face poorer outcomes. BRCA2 carriers were also found to have a higher risk of progression to castration-resistant prostate cancer. These findings highlight the importance of evaluating germline mutations among prostate cancer patients. Learn more by reading the full article at: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post93024/
ICARE Social Media Post September 2024
NCCN CEG Guideline Update #3: Use of HRT following premature surgical menopause
The National Comprehensive Cancer Network (NCCN) released the new Genetic/Familial High-Risk Assessment Colorectal, Endometrial, and Gastric Cancer guidelines on August 8th, 2024. Updates include use of hormone replacement treatment following premature surgical menopause from risk-reducing oophorectomy. You can check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_ceg.pdf
Permanent link to this article: https://inheritedcancer.net/poat92524/
ICARE Social Media Post September 2024
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update – VERSION 3
The National Comprehensive Cancer Network (NCCN) released the new Genetic/Familial High-Risk Assessment Colorectal, Endometrial, and Gastric Cancer guidelines on August 8th, 2024. Updates include gynecologic risk and preventive surgery considerations for MLH1, MSH6, MSH2, and PMS2 carriers. You can check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_ceg.pdf
Permanent link to this article: https://inheritedcancer.net/post91824/
ICARE Social Media Post September 2024
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update – VERSION 2
The National Comprehensive Cancer Network (NCCN) released the new Genetic/Familial High-Risk Assessment Colorectal, Endometrial, and Gastric Cancer guidelines on August 8th, 2024. Updates include ⤸ You can check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_ceg.pdf
Permanent link to this article: https://inheritedcancer.net/post91424/
ICARE Social Media Post September 2024
NCCN Breast, Ovarian, and Pancreatic Cancer Guidelines Update
The National Comprehensive Cancer Network (NCCN) released updated Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer guidelines on September 11th, 2024. Some updates include ⤸ To read more, you can check out the full guidelines by creating a FREE account at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf
Permanent link to this article: https://inheritedcancer.net/post91224/
ICARE Social Media Post September 2024
NCCN Colorectal, Endometrial, and Gastric Cancer Guidelines Update
The National Comprehensive Cancer Network (NCCN) released the new Genetic Familial High-Risk Assessment Colorectal, Endometrial, and Gastric Cancer guidelines on August 8th, 2024. Updates include ⤸• Endometrial cancer recommendations included throughout• Hereditary Diffuse Gastric Cancer section added (HGAST-1)• Gynecologic risk and preventive surgery considerations for those with Lynch Syndrome• Use of hormone replacement treatment following …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post90324/
ICARE Social Media Post August 2024
ICARE Overview Video
Learn more about the Inherited Cancer Registry (ICARE) by watching an exciting presentation by student researcher, Reagan Sellers, who provides information about the history of ICARE, what being an ICARE participant entails, our various education and engagement efforts, and the focused research studies being conducted through ICARE. Check out the full video on our YouTube …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post82824/
ICARE Social Media Post August 2024
BRCA1/2 carriers: Melanoma Risk?
A new study in which ICARE participants were included reported that compared to 1.5% in the general population, the risk of melanoma may be slightly elevated: in BRCA1/2 carriers:○ BRCA1: 2.5%○ BRCA2: 2.3% Read the full article to learn more at: https://tinyurl.com/ye2xxfru Reference: Narod et al. Hered Cancer Clin Pract. 2024;22(1):7. PMID: 38741145.
Permanent link to this article: https://inheritedcancer.net/post82624/
ICARE Social Media Post August 2024
Peritoneal Cancer After Oophorectomy in BRCA1/2 Carriers
A recent study among BRCA1 and BRCA2 carriers, which included ICARE participants, found that the risk for peritoneal cancer following a preventive bilateral oophorectomy was higher among BRCA1 carriers than BRCA2 carriers. Specifically, among 6310 women, the annual risk of peritoneal cancer was 0.14% for BRCA1 carriers and 0.06% for BRCA2 carriers, and the 20-year …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post82524/
ICARE Social Media Post August 2024
How to Access NCCN Guidelines
Unlock valuable resources for your health journey! 🌟 The National Comprehensive Cancer Network (NCCN) guides clinical practice by providing free access to their guidelines that are available to both patients and healthcare providers. To access the most recent version of cancer-related guidelines that are updated at least annually, create a free username and password at …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post82024/
ICARE Social Media Post August 2024
FDA Approved a Blood Test for Colorectal Cancer Screening
Breaking News: The FDA recently approved a blood test for colorectal cancer screening! 🔍 What does this mean? ⭐ Bottom Line: Learn more at: https://www.nejm.org/doi/full/10.1056/NEJMoa2304714 Reference: Chung, et al. N Engl J Med. 2024;390(11):973-983: PMID: 38477985.
Permanent link to this article: https://inheritedcancer.net/post80824/
ICARE Social Media Post August 2024
Comparing Cancer Risk Management between Females with Truncating CHEK2 1100delC versus Missense CHEK2 I157T Variants
In a recent study based on ICARE participants, we compared cancer risk management between those with CHEK2 1100delC (which leads to moderate breast cancer risk, generally more than 20% lifetime risk) versus I157T (which is a low penetrance mutation, with breast cancer risks of less than 20%) 👩🔬 Of the 150 CHEK2 carriers, there were …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post80524/
ICARE Social Media Post July 2024
Genetics Role in Identifying Lower Breast Cancer Risk
A new study of 25,591 women found: Based on genetic risk, identifying individuals at: This study challenges a one-size-fits all mammogram screening approach. This approach could possibly reduce rising healthcare costs by reallocating resources and lower unnecessary anxiety that comes with frequent screenings, and even prevent unnecessary procedures. Read the full article to learn more …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post72924/
ICARE Social Media Post July 2024
New Study in POT1 Carriers
In recognition of Sarcoma Awareness Month, a new study found that POT1 mutations are associated with an increased risk for melanoma and a wider variety of different sarcomas than previously described. Specifically, the study showed significant raised risks of around 7-fold for both sarcoma and melanoma. This included many different types of sarcomas, including: Learn …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post72824/
ICARE Social Media Post July 2024
New Genes Raise Breast Cancer Risks Identified in Study of African Women
A new study, which included ICARE-related efforts through including BEST study participants:▪️ Identified 12 breast cancer genes that may help to predict breast cancer risk▪️ Included more than 40,000 women of African ancestry, almost half of which had breast cancer Why is studying individuals from diverse ancestral backgrounds important?▪️ Refine risks across diverse populations▪️ A …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post72524/
ICARE Social Media Post June 2024
ICARE Post #3 – PALB2 Genomics Study Post
If you have a history of breast cancer and a mutation in the PALB2 gene, you may be eligible to contribute to tumor genomics studies to help better understand inherited breast cancer in PALB2 carriers. Through the Inherited Cancer Registry (ICARE), we are studying: With your help, we hope that one day our research may …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post71124/
ICARE Social Media Post June 2024
ICARE Post #3 – CHEK2 Genomics Study Post
If you have a history of breast cancer and a mutation in the CHEK2 gene, you may be eligible to contribute to tumor genomics studies to help better understand inherited breast cancer in CHEK2 carriers. Through the Inherited Cancer Registry (ICARE), we are studying: With your help, we hope that one day our research may …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post7052024/
ICARE Social Media Post June 2024
ICARE Post #3 – ATM Genomics Study Post
If you have a history of breast cancer and a mutation in the ATM gene, you may be eligible to contribute to tumor genomics studies to help better understand inherited breast cancer in ATM carriers. Through the Inherited Cancer Registry (ICARE), we are studying: With your help, we hope that one day our research may …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post62324/
ICARE Social Media Post June 2024
Breast Cancer Patients with BRCA Variants Diagnosed Within 10 Years Postpartum at Higher Mortality Risk
A new study reported that among BRCA carriers, breast cancer diagnosed within 10 years of having a child was associated with a higher risk of mortality, especially in BRCA1 carriers. This information could be important for genetic counseling, prevention, and treatment strategies in BRCA carriers. Learn more by reading the full article at: https://bit.ly/3wHyAIz Reference: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post60624/
ICARE Social Media Post June 2024
Pregnancy after Breast Cancer: BRCA carriers
A new study reported that pregnancy after breast cancer was not associated with adverse maternal prognosis or fetal outcomes. These findings suggest that pregnancy after breast cancer is safe for both mother and baby. Read the full article at: https://bit.ly/3wcoa3xReference: Newman, et al. JAMA Surg. 2024. Online ahead of print. PMID: 38536201. Follow us to …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post60324/
ICARE Social Media Post May 2024
New ASCO Guideline: Selection of Germline Testing Panels in Patients With Cancer
The American Society of Clinical Oncology (ASCO) just released new guidelines for the selection of germline genetic testing panels in patients with cancer. Items addressed include ⤸ What elements are most important in the collection of family history?🧬 Details of cancers in first- and second-degree relatives and the patient’s ethnicity. When and how to use …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post52124/
ICARE Social Media Post May 2024
AACR Cancer Disparities Progress Report 2024
The American Association for Cancer Research (AACR) just released a Cancer Disparities Progress Report on May 15th, which includes highlights on inherited cancers! On Page 163, this report discusses policies to address disparities in screening and surveillance for hereditary cancer syndromes. It also highlights an electronic support tool, called MyLynch, that can be used to …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post5172024/
ICARE Social Media Post May 2024
CHEK2: Bi-Allelic Mutations
Do you know the cancer risks for individuals with CHEK2 bi-allelic mutations (i.e., those with TWO CHEK2 mutations)? A new study found that risks may be higher for multiple types of cancer, including breast cancer, in both females and males ⤸▪️ Females more frequently developed 2 or more cancers at younger ages▪️ Males more frequently …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post50324/
ICARE Social Media Post April 2024
YouTube Channel
Curious about inherited cancer and the latest breakthroughs? Look no further than the ICARE YouTube channel! 🎥 Join genetics experts as they dive into discussions on important topics like updates to NCCN cancer risk management guidelines and insights into specific inherited cancer genes. 💡 Don’t miss out on some of our top-viewed videos covering management …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post42824/
ICARE Newsletter Spring 2024
Inherited Risk in Patients with Pancreatic Acinar Cell Carcinoma
In a study of a rare type of pancreatic cancer, called pancreatic acinar cell cancer (PACC), over one third (36.7%) of a total of 49 patients with PACC had a mutation in an inherited cancer gene. The most commonly mutated gene was BRCA2 (12), and other genes included BRCA1 (1), PALB2 (2), ATM (2), and …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2024-inherited-risk-in-patients-with-pancreatic-acinar-cell-carcinoma/
ICARE Newsletter Spring 2024
BRCA-Associated Prostate Cancer Treatment Updates
New studies to guide treatment strategies in men with prostate cancer and a BRCA mutation were recently published. Specifically, a recent study suggested that platinum-based chemotherapy may be as effective as PARP inhibitors for individuals with BRCA-positive metastatic castration-resistant prostate cancer.1 The study sheds light on treatment options for advanced prostate cancer patients. More recently, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2024-brca-associated-prostate-cancer-treatment-updates/
ICARE Newsletter Spring 2024
Ask the Expert
This question was addressed by Ronald D. Alvarez, MD, MBA, Professor and Chairman of the Department of Obstetrics and Gynecology at the Vanderbilt University Medical Center in Nashville, Tennessee. He is also the current vice chair of the National Comprehensive Cancer Network (NCCN) Ovarian Cancer Treatment Guidelines and has served in multiple leadership roles in …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2024-ask-the-expert/
ICARE Newsletter Spring 2024
How Well Do Cancer Risk Management strategies Work Among BRCA Carriers
Several important studies were published recently on the effectiveness of risk management strategies in BRCA carriers. Specifically, a recently published study in which ICARE participants were included suggested that preventive bilateral mastectomy for BRCA carriers greatly reduced the risk of developing breast cancer by 80%.1 Additionally, study findings showed that after preventive mastectomy, the chance …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2024-how-well-do-cancer-risk-management-strategies-work-among-brca-carriers/
ICARE Newsletter Spring 2024
Aerobic Exercise May Reduce Risk of Colorectal Cancer in Patients with Lynch Syndrome
A new research study among individuals with Lynch Syndrome reported that regular and intense aerobic exercise may lower the risk of colorectal cancer by improving the immune system’s ability to detect and remove potentially harmful cells. Specifically, the researchers enrolled 21 patients with Lynch Syndrome either to an 1) exercise group (45 minute cycling classes …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2024-aerobic-exercise-may-reduce-risk-of-colorectal-cancer-in-patients-with-lynch-syndrome/
ICARE Newsletter Spring 2024
New Guidelines Released Through ASCO-Society of Oncology; Germline Testing in Patients with Breast Center
In January 2024, the American Society of Clinical Oncology (ASCO) in conjunction with the Society of Surgical Oncology released new guidelines for germline testing in patients with breast cancer, which include the following: For a full list of recommendations in this guideline, the article is available at: https://ascopubs.org/doi/10.1200/JCO.23.02225 Bedrosian, et al. J Clin Oncol. 2024;42(5):584-604. PMID:38175972. Social …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2024-new-guidelines-released-through-asco-society-of-oncology-germline-testing-in-patients-with-breast-center/
ICARE Newsletter Spring 2024
National Comprehensive Cancer Network (NCCN) Guideline Updates
Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Cancer – Released February 12th, 2024 (V3.2024) Check out the full guidelines by creating a FREE account at www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf Contralateral breast cancer risks in these updated guidelines: Expanded guidance about gynecologic cancers in BRCA1/2 carriers: Some highlights related to HRT include: Genetic/Familial High-Risk Assessment: Colorectal Cancer – Released …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2024-national-comprehensive-cancer-network-nccn-guideline-updates/
ICARE Social Media Post March 2024
Risk Management Strategies for Women with BRCA1/2
Two new research studies that 𝗶𝗻𝗰𝗹𝘂𝗱𝗲𝗱 𝗜𝗖𝗔𝗥𝗘 𝗽𝗮𝗿𝘁𝗶𝗰𝗶𝗽𝗮𝗻𝘁𝘀 showed that risk of death is lowered among BRCA1/2 carriers with 1) MRI screening for breast cancer and 2) removal of both ovaries and fallopian tubes. 💡 In the first study, MRI screening greatly lowered the risk of death from breast cancer (hazard ratio of 0.23) with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post3524/
ICARE Social Media Post February 2024
Risk-reducing mastectomy and breast cancer mortality in women with BRCA1/BRCA2
🔬 Exciting news! A recent study, which included ICARE participants, suggests the life-saving potential of preventive mastectomy for BRCA1 and BRCA2 carriers. 💪 This procedure significantly reduces the risk of developing breast cancer and may even decrease the risk of breast cancer-related mortality. The study found that after preventive mastectomy, the chance of dying from …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post22724/
PARP Inhibitor (Olaparib) in men with BRCA mutations and prostate cancer
A recent study found that Olaparib (Lynparza) improved survival outcomes among men with BRCA1/2 mutations and metastatic castration-resistant prostate cancer, regardless of whether the mutation was germline or somatic. This underscores the potential of targeted therapies in improving outcomes for those with inherited cancer gene mutations. Learn more at: https://ascopubs.org/doi/10.1200/JCO.23.00339 Reference: Mateo, et al. J …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post22024/
ICARE Social Media Post February 2024
Updates to NCCN Guidelines: Genetic/Familial Breast, Ovarian, and Pancreatic Post #1
The National Comprehensive Cancer Network (NCCN) just released updated Genetic/Familial Breast, Ovarian, and Pancreatic Cancer guidelines on February 12th, 2024! Updates include adding contralateral breast cancer risks for BRCA1, BRCA2, PALB2, CHEK2, and other genes to the GENE-A (Cancer Risk Management) table 🧬 You can check out the full guidelines by creating a FREE account …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post21324/
ICARE Social Media Post February 2024 Updates to NCCN Guidelines: Genetic/Familial Breast, Ovarian, and Pancreatic Post #2
The National Comprehensive Cancer Network (NCCN) just released updated Genetic/Familial Breast, Ovarian, and Pancreatic Cancer guidelines on February 12th, 2024! Updates include expanded guidance about gynecologic cancers in BRCA1 and BRCA2, including:✓ Reproductive considerations✓ Non-surgical and surgical risk reduction✓ Salpingectomy✓ Hysterectomy considerations✓ HRT after risk-reducing removal of the ovaries You can check out the full …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post21324_2/
ICARE Social Media Post February 2024
Germline EGFR Mutations and Familial Lung Cancer
As featured in our Fall 2023 newsletter, inherited T790M EGFR mutations lead to a higher risk of lung cancer and are more common in the Southeast United States due to a ‘founder effect’. A study, in which our fellow Vanderbilt colleagues contributed, among individuals with this inherited cancer gene mutation found that more than half …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post21224/
ICARE Social Media Post January 2024
Hereditary Cancer Genetic Testing Educational Video
Want to learn more about genetic testing for inherited cancer? We use patient stories to give information about testing, its possible value to individuals and their family, and ways to access resources to learn more about getting tested. Check out the video here: http://tinyurl.com/genetictestingvideo To get free personalized information about hereditary cancer risk and testing, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11724/
ICARE Social Media Post January 2023
Colon Adenomas in Lynch Syndrome
New research reveals that individuals under the age of 50 with Lynch Syndrome often develop small, flat adenomas, particularly in the right (proximal) colon. This finding emphasizes the importance of rigorous screening, with a special focus on the proximal colon, among these young Lynch Syndrome patients 🩺🔍 Learn more at: https://www.dldjournalonline.com/article/S1590-8658(23)00946-5/fulltext Reference: Alric, et al. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11024/
ICARE Social Media Post January 2023
New ASCO Germline Testing in Patients With Breast Cancer Guidelines
The American Society of Clinical Oncology (ASCO) just released new guidelines for germline testing in patients with breast cancer, which include the following:🧬 BRCA1/2 testing should be offered to ALL patients diagnosed with breast cancer at or below age 65🧬 Testing for other hereditary cancer genes should also be offered based on personal and family …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post10524/
ICARE Social Media Post January 2023
BRCA/Prostate Cancer/Treatment
A recent study suggests that Platinum Chemotherapy is as effective as PARP inhibitors for individuals with BRCA-positive metastatic castration-resistant prostate cancer. The study sheds light on treatment options for advanced prostate cancer patients 🩺✨ Learn more at: http://tinyurl.com/3vs2mk8f Reference: Fazekas, et al. Eur Urol Oncol. 2023: S2588-9311(23)00174-8. PMID: 37722977.
Permanent link to this article: https://inheritedcancer.net/post10324/
ICARE Social Media Post December 2023
BRCA1/2 carriers with Risk Reducing Salpingo-oophorectomy
A study among BRCA1/2 carriers who underwent a risk-reducing salpingo-oophorectomy (i.e., removal of both ovaries and fallopian tubes) found that the risk of peritoneal cancer increases if serous tubal intraepithelial carcinoma (STIC) is present – specifically, peritoneal cancer risk was 10.5% with STIC versus 0.3% without STIC at 5 years and 27.5% with STIC versus …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post121923/
ICARE Social Media Post December 2023
BRCA1/2: removing both ovaries and tubes to lower risks
A recent study among nearly 500 BRCA1/2 carriers who underwent breast cancer surgery found that survival was higher in those who chose preventive removal of their ovaries and fallopian tubes (in order to reduce cancer risks) compared to those who did not, particularly in BRCA1 carriers. Learn more at: https://tinyurl.com/5hdx6ytv Reference: Martelli, et al. JAMA …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post121323/
ICARE Social Media Post December 2023
Tamoxifen and Breast Cancer Risk in BRCA Mutations
In a recent study among female BRCA1/2 carriers, including ICARE participants, researchers explored the impact of using tamoxifen and/or raloxifene on breast cancer risk. 👩🔬 After almost 7 years of follow-up, only 10.9% of those in the tamoxifen/raloxifene group were diagnosed with breast cancer, compared to 14.3% of those in the non-user group. These findings …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post120523/
ICARE Social Media Post November 2023
Aerobic Exercise May Be Effective At Reducing Risk Of Colorectal Cancer In Patients With Lynch Syndrome
New research suggests regular, intense aerobic exercise may lower colorectal cancer risk in Lynch Syndrome patients 🏃♂️🧬 Learn more at: https://tinyurl.com/2s44828h Reference: Deng, et al. Clin Cancer Res. 2023; 29(21): 4361-4372. PMID: 37724990.
Permanent link to this article: https://inheritedcancer.net/post111523/
ICARE Social Media Post November 2023
Inherited Genes Associated With Aggressive Types Of Prostate Cancer
A new study found that inherited BRCA2, ATM, and NBN genes were most strongly associated with aggressive or metastatic prostate cancer. Learn more at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2809874 Reference: Darst, et al. JAMA Oncol. 2023. PMID: 37733366.
Permanent link to this article: https://inheritedcancer.net/post110923/
ICARE Social Media Post November 2023
Piloting a Spanish-Language Web-Based Tool for Hereditary Cancer Genetic Testing
Language barriers should never prevent people from accessing essential information in the field of healthcare. To fill the gap caused by the lack of Spanish-speaking genetic counselors, our team developed a web-based genetic education tool 🧬 A study among 41 Spanish-speaking found that viewing the tool significantly increased knowledge and feeling informed and empowered to …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post110123/
ICARE Social Media Post November 2023
Piloting a Spanish-Language Web-Based Tool for Hereditary Cancer Genetic Testing
Las barreras del idioma nunca deberían impedir que las personas accedan a información esencial en el campo de la atención de salud. Para llenar el vacío causado por la falta de asesores genéticos que hablan español, nuestro equipo desarrolló una herramienta de educación genética basada en la web. Un estudio entre 41 hispanohablantes encontró que …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post110123_1/
Permanent link to this article: https://inheritedcancer.net/post102923/
ICARE Social Media Post October 2023
Triple-negative breast cancers across populations
Triple-negative breast cancers, which do not have estrogen, progesterone, or HER2 receptors, can be more serious and difficult to treat. Inherited breast cancer gene mutations, like BRCA1/2, are more common among this type of breast cancer – which is why it is important for those with triple-negative breast cancer to consider getting genetic testing that …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102223/
ICARE Social Media Post October 2023
BRCA1/2 Carriers with Risk-Reducing Salpingo-Oophorectomy: Cancer Worry?
A study found that the majority of BRCA1/2 carriers who underwent risk-reducing salpingo-oophorectomy have declining cancer worry. However, a subset had concerns – these individuals are important to identify and try to offer additional support to. Use the following link to learn more: https://bit.ly/3qflyig Reference: van Bommel, et al. Support Care Cancer. 2022;30(4):3409-3418. PMID: 34997316.
Permanent link to this article: https://inheritedcancer.net/post102023/
ICARE Social Media Post October 2023
Navigating BRCA1/2 Choices
A recent randomized controlled trial among 107 BRCA1/2 carriers, which included ICARE participants, evaluated the effectiveness of a behavioral phone intervention delivered by genetic counselors on the uptake of risk-reducing salpingo-oophorectomy. Women who received the intervention had significantly lower decisional conflict and higher knowledge after one year, and at the two year mark, nearly 54% …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post101223/
ICARE Social Media Post October 2023
BRCA2 & Melanoma Risk
Are BRCA2 carriers at higher risk for developing melanoma? As featured in our Fall 2023 newsletter, at present, we are not sure because results from different studies have been inconsistent. For instance, one study among 173 families with a BRCA2 mutation found a 2.6-fold risk of developing melanoma; however, another study of 139 Dutch families …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post100623/
ICARE Newsletter Fall 2023
Community Spotlight
My paternal grandparents were my heroes. Wise beyond their time, they relished teaching our familythat knowledge is power, health is everything, and love is unconditional. Back then, Prevention healthmagazine and vitamin supplements filled their mailbox and 1960’s exercise guru Jack LaLane, and health foodadvocate Euell Gibbons, beckoned new followers from a talking picture box in …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-community-spotlight/
ICARE Newsletter Fall 2023
Ask the Expert
This question was addressed by Kelly Taylor, MS, LCGC, a licensed and certified genetic counselor with expertise inresearch genetics and hereditary cancers at the Vanderbilt Hereditary Cancer Clinic. If you have a question you wouldlike addressed, please email the ICARE team at ICARE@vumc.org for consideration in future newsletters. Q: I have Lynch Syndrome and an …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-ask-the-expert/
ICARE Newsletter Fall 2023
BRCA-associated Prostate Cancers
On April 28th, 2023, the FDA approved olaparib plus abiraterone acetate for first line treatment for metastatic castration-resistant prostate cancer, but only in patients whose tumors have BRCA mutations. Although a broad indication for the combination therapy was desired, concerns about the trial design were raised, and the phase III results did not explicitly show …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-brca-associated-prostate-cancers/
ICARE Newsletter Fall 2023
Healthcare Delivery
A recent study, based on ICARE participants, found that getting care according to guidelines depends on what the healthcare provider recommends, as well as how much trust the patient has in their care. This study shows us how important it is to find ways to improve knowledge among healthcare providers and trust in care among …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-healthcare-delivery/
ICARE Newsletter Fall 2023
Germline EGFR Mutations and Familial Lung Cancer
A study, in which our Vanderbilt colleagues Georgia Wiesner, MD, MS (geneticist) and Kelly Taylor, MS, LCGC (genetic counselor) participated, was recently published about the inherited T790M EGFR mutation. Mutations in this gene lead to a higher risk or lung cancer and were found to be more common in the Southeast United States where there …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-germline-egfr-mutations-and-familial-lung-cancer/
ICARE Newsletter Fall 2023
Genes Associated with Aggressive Prostate Cancer
A new study of almost 18,000 men with prostate cancer showed that inherited mutations in the BRCA2, ATM, and NBN genes were strongly associated with aggressive prostate cancer. Less strong associations were seen for inherited mutations in the MSH2, XRCC2, and MRE11A genes. The findings of this study suggest that knowing about inherited genes that …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-genes-associated-with-aggressive-prostate-cancer/
ICARE Newsletter Fall 2023
Are BRCA2 Carriers at Higher Risk for Melanoma?
At present, we are not sure because results from different studies have been inconsistent (1). For instance, one study among 173 families with a BRCA2 mutation found a 2.6-fold risk of developing melanoma (2); however, another study of 139 Dutch families with a BRCA2 mutation revealed a lower than general population risk for melanoma (3). …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-are-brca2-carriers-at-higher-risk-for-melanoma/
ICARE Newsletter Fall 2023
Fertility Treatment in BRCA Carriers: Breast Cancer Risk
A recent study among female BRCA carriers showed risk of breast cancer was not significantly raised through fertility treatment, which is reassuring. There remains a need to further study this question in more detail, for associations with breast cancers across different breast cancer subtypes, including breast cancers that are hormone-related and breast cancers that are …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-fertility-treatment-in-brca-carriers-breast-cancer-risk/
ICARE Newsletter Fall 2023
Newly released ACMG Clinical Practice Resource on CHEK2 Developed Through a Group of Worldwide Experts!
A person with a pathogenic variant in the CHEK2 gene may be at an increased risk for developing breast and other cancers. This ACMG Clinical Practice Resource, published in ACMG’s flagship journal, Genetics in Medicine, provides valuable information for healthcare providers caring for individuals with pathogenic variants in the CHEK2gene. This new ACMG Practice Resource …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-newly-released-acmg-clinical-practice-resource-on-chek2-developed-through-a-group-of-worldwide-experts/
ICARE Newsletter Fall 2023
Tamoxifen and Breast Cancer Risk in Women with a BRCA1 or BRCA2 Mutation
In a study of female BRCA carriers, including ICARE participants, those that used tamoxifen and/or raloxifenewere compared to those that did not. After an average follow-up time of almost 7 years, 10.9% of those in the tamoxifen/raloxifene group were diagnosed with breast cancer, compared to 14.3% in the group that did not use thesedrugs. This …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-tamoxifen-and-breast-cancer-risk-in-women-with-a-brca1-or-brca2-mutation/
ICARE Newsletter Fall 2023
BRCA1 and BRCA2 Carriers: Cancer Risks with Oral Contraceptive Use (UK)
Among female BRCA1 and BRCA2 carriers, a recent study found that oral contraceptive use is associated with:› Raised risk of breast cancer, but only in those using for over 5 years (relative risk: 1.25)› Lower risk of ovarian cancer (nearly cut in half) Park, et al. Carcinogenesis. 2022;43(3):231-242. PMID:
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-brca1-and-brca2-carriers-cancer-risks-with-oral-contraceptive-use-uk/
ICARE Newsletter Fall 2023
Recommendations for Inherited Ovarian Cancer Genes: United Kingdom (UK)
Through consensus, management recommendations were developed in the UK for females with pathogenic variants inthe following inherited ovarian cancer genes: RAD51C, RAD51D, BRIP1, and PALB2. Hanson, et al. J Med Genet. 2023;60(5):417-429. PMID: 36411032. Social media post September 26th, 2023.Available at https://tinyurl.com/post9262023.
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-recommendations-for-inherited-ovarian-cancer-genes-united-kingdom-uk/
ICARE Newsletter Fall 2023
Mammograms: New U.S. Preventive Services Task Force (USPSTF) Recommendations
On May 9th, 2023, the USPSTF published a new draft recommendation for all cisgender women and those assigned female sex at birth to do mammograms from ages 40 to 74, every two years, for those at average (population) risk forbreast cancer. This change in recommendation is due to recent troubling trends, including an increase in …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-mammograms-new-u-s-preventive-services-task-force-uspstf-recommendations/
ICARE Newsletter Fall 2023
National Comprehensive Cancer Network (NCCN) Guidelines Updates
Check out the full NCCN guidelines by creating a FREE account at www.nccn.org Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic CancerReleased August 28th, 2023 (V1.2024) › Transgender, Non-Binary, and Gender Diverse Individuals: NEW section on care (Page 63-66, TNBGD-1 to 4)› Li-Fraumeni Syndrome: Significant updates to content (risks and care) (Pages 57-60, LIFR-A): Table added …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2023-national-comprehensive-cancer-network-nccn-guidelines-updates/
ICARE Social Media Post October 2023
What Are Established Cancer Genes?
There are many different inherited breast cancer genes, and different genes lead to different levels of breast cancer risk:
Permanent link to this article: https://inheritedcancer.net/post100423/
ICARE Social Media Post August 2023
BRCA1/2 Outcomes: Metastatic Breast Cancer
According to a recent study, germline BRCA1/2 status did not affect prognosis. However, outcomes differed by immunohistochemistry (IHC) subtype as follows: Use the link in bio to learn more! Reference: Mailliez, et al. Int J Cancer. 2023;152(5):921-931. PMID: 36161271.
Permanent link to this article: https://inheritedcancer.net/post83032/
ICARE Social Media Post August 2023
Updates to NCCN Guidelines: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic
The National Comprehensive Cancer Network (NCCN) just released updated Genetic/Familial Breast, Ovarian, and Pancreatic Cancer guidelines on August 28th, 2023, which included: You can check out the full guidelines by creating a FREE account at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf
Permanent link to this article: https://inheritedcancer.net/post82923/
ICARE Social Media Post August 2023
FDA Approval Post – Approves Niraparib
A critical step forward in cancer care! The FDA has approved the use of niraparib and abiraterone acetate with prednisone in treating patients with BRCA-mutated castration-resistant prostate cancer. The green light comes backed by the robust efficacy data from the MAGNITUDE trial. Read more about the FDA approval at https://tinyurl.com/dzdvtm9m Learn more about the MAGNITUDE …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post82423/
ICARE Social Media Post August 2023
Cancer Risks: Inherited CDH1 Mutations
A recent study found that inherited CDH1 mutations: These findings support expansion of lobular breast cancer-centered criteria. Use this link to learn more: http://bit.ly/3DJa7Cs Reference: Garcia-Pelaez, et al. Lancet Oncol. 2023 Jan;24(1):91-106. PMID: 36436516.
Permanent link to this article: https://inheritedcancer.net/post82223/
ICARE Social Media Post August 2023
UK Recommendations for: Inherited Ovarian Cancer Genes
Through consensus, management recommendations were developed in the UK for females with pathogenic variants in the following inherited ovarian cancer genes: Learn more at the link in bio! Reference: Hanson H, et al. J Med Genet. 2023;60(5):417-429. PMID: 36411032.
Permanent link to this article: https://inheritedcancer.net/post81823/
ICARE Social Media Post August 2023
Fertility Treatment in BRCA Carriers: Breast Cancer Risk
A study found that fertility treatment did not significantly raise the risk of breast cancer, which is reassuring. However, further research is required with regard to: Use the link in bio to learn more! Reference: Liu et al. Front Endocrinol (Lausanne). 2022;13:986477. PMID: 36176466.
Permanent link to this article: https://inheritedcancer.net/post81123/
ICARE Social Media Post August 2023
New Article – Cancer Risk Management
A recent study, which 𝗶𝗻𝗰𝗹𝘂𝗱𝗲𝗱 𝗜𝗖𝗔𝗥𝗘 𝗽𝗮𝗿𝘁𝗶𝗰𝗶𝗽𝗮𝗻𝘁𝘀, found that getting care according to guidelines depends on: Therefore, we need ways to improve knowledge among healthcare providers and trust in care among patients! Use this link to learn more: https://tinyurl.com/n26m4zys Reference: Dean, et al. Genet Med. 2023;100945. PMID: 37515473.
Permanent link to this article: https://inheritedcancer.net/post80723/
ICARE Social Media Post July 2023
ACMG Clinical Practice Resource on CHEK2
Check out the newly released ACMG Clinical Practice Resource on CHEK2 developed through a group of worldwide experts! 👇https://tinyurl.com/bp8esxsf Read the ACMG press release at 👇https://tinyurl.com/yvammhzt
Permanent link to this article: https://inheritedcancer.net/post72623/
ICARE Social Media Post July 2023
Inherited Cancer Genes in Children: BRCA1/2, PALB2, ATM, CHEK2 , Lynch Genes
Recent study findings suggest that BRCA1/2, PALB2, ATM, CHEK2, and the Lynch Syndrome genes might confer reduced penetrance cancer risk among children. However, there are no adjustments to management or testing recommendations based on the level of risk (i.e., normally do not test children for conditions that primarily increase the risk of cancer in adulthood). …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post72123/
ICARE Social Media Post July 2023
BRCA1/2 carriers: Risks with Oral Contraceptives Use
Among female BRCA1 and BRCA2 carriers, a recent study found that oral contraceptive use is associated with an increased risk of breast cancer, but only among users who have been using them for more than five years, while ovarian cancer risk was nearly cut in half. Use the link in bio to learn more! Reference: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post70623/
ICARE Social Media Post July 2023
CDKN2A Mutations: Pancreatic Cancer Screening over 20 years of follow up
A study among CDKN2A carriers assessing the yield and outcomes of 20 years of pancreatic cancer screening found: These results highlight the potential importance of pancreatic cancer screening among CDKN2A carriers. Currently, NCCN guidelines recommend annual pancreatic cancer screening starting at age 40, regardless of family history. Use the link in bio to learn more! …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post70223/
ICARE Social Media Post June 2023
Breast Cancer Risk Model: Predicting Risk over 10-year period
A new study found that mammographic features (density, microcalcifications, masses, and left-right breast asymmetries) predicted risks better than traditional lifestyle/familial risk-based risk models over a 10-year period. Use this link to learn more: https://bit.ly/443GDLd Reference: Eriksson et al. J Clin Oncol. 2023;JCO2201564. PMID: 36930854.
Permanent link to this article: https://inheritedcancer.net/post62823/
ICARE Social Media Post June 2023
Li-Fraumeni Syndrome/TP53 mutations
A recent study found that, regardless of a prior cancer diagnosis, there was no increased cancer worry after 1 year of full-body MRI surveillance in TP53 carriers. Further research is needed to evaluate these issues further. Use the link in bio to read the full article! Reference: Omran, et al. Cancer. 2023;129(6):946-955. PMID: 36601958.
Permanent link to this article: https://inheritedcancer.net/post62123/
ICARE Social Media Post June 2023
Should Black women be screened earlier?
A recent study found that Black women had the highest rate of breast cancer deaths in their 40s, compared to White women and women of other races and ethnicities. Use this link to learn more: https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2803948?utm_source=For_The_Media&utm_medium=referral&utm_campaign=ftm_links&utm_term=041923 Reference: Chen T, et al. JAMA Netw Open. 2023;6(4):e238893. PMID: 37074714.
Permanent link to this article: https://inheritedcancer.net/post62023/
ICARE Social Media Post June 2023
FDA Advisory Committee Recommendation: Olaparib for Prostate Cancer Treatment
Olaparib plus abiraterone acetate is recommended as the first line treatment for metastatic castration-resistant prostate cancer, but only in patients whose tumors have BRCA mutations. Although a broad indication for the combination therapy was desired, concerns about the trial design were raised, and the phase III results did not explicitly show that patients without a …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post61623/
Permanent link to this article: https://inheritedcancer.net/post62223/
ICARE Social Media Post June 2023
Updates to NCCN Guidelines: Genetic/Familial High-Risk Assessment: Colorectal Version 1.2023
The National Comprehensive Cancer Network (NCCN) just released updated colorectal cancer guidelines which includes: You can check out the full guidelines by creating a FREE account at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf
Permanent link to this article: https://inheritedcancer.net/post60823/
ICARE Social Media Post June 2023
Inherited Gene Mutation in Those with Multiple Primary Cancers
Among patients with multiple primary cancers, the proportion with inherited cancer gene mutations increased with the number of primary cancers:• 2 cancers: 13.1%• 3 cancers: 15.9%• 4+ cancers: 18.0% Many of the double primaries, such as those of the breast, ovary, colorectum, and endometrium, occurred in women who would already be eligible for genetic testing. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post60723/
ICARE Social Media Post June 2023
June 4th: National Cancer Survivor’s Day
Today is National Cancer Survivors Day! Learn how you can support cancer survivors by visiting the American Association for Cancer Research (AACR) website 👇https://www.aacr.org/patients-caregivers/awareness-months/national-cancer-survivor-month/
Permanent link to this article: https://inheritedcancer.net/post60523-2/
ICARE Social Media Post June 2023
VUS Results: Rates of Reclassification in Inherited Cancer Genes
In a multicenter study evaluating reclassifications of variant of uncertain significance (VUS) results in breast, ovarian, and colorectal cancer susceptibility genes, it was discovered that of 3261 VUS results, 8.1% were reclassified. Of all the reclassified VUS results, 11.3% resulted in clinically actionable findings, and 4.6% led to changes in clinical management. The reclassification rates …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post60523/
ICARE Social Media Post May 2023
Cancer Care: Transgender and Gender-Diverse Persons
Did you know that transgender and gender-diverse persons face unique challenges that can influence cancer risk and outcomes? For instance, these individuals face barriers to healthcare access and inequities in treatment, with healthcare providers lacking knowledge about the health needs of this population. Solutions are needed to offer the best care for these individuals. Use …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post52223/
ICARE Social Media Post May 2023
Women’s Health Week
In honor of National Women’s Health Week from May 14th to May 20th, we would like to encourage you to speak with your healthcare provider to make sure you are up-to-date on your cancer screenings, such as mammograms and colonoscopies, to help prevent cancer or find it early when it is easier to treat. Spread …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post51523/
ICARE Social Media Post May 2023
ATM breast cancer risks: High Risk c.7271T>G mutation
According to a recent study, while there was a reported 2-fold risk for breast cancer overall among ATM carriers, those with the c.7271T>G mutation had the highest risk. Use the link to learn more: https://bit.ly/40bKovj Reference: Hall et al. Cancer Prev Res (Phila). 2021;14(4):433-440. PMID: 33509806.
Permanent link to this article: https://inheritedcancer.net/post51123/
ICARE Social Media Post May 2023
USPSTF New Recommendation: Mammograms Start at Age 40
Yesterday the United States Preventive Services Task Force (USPSTF) published a new draft recommendation for all cisgender women and those assigned female sex at birth to do mammograms from ages 40 to 74, every two years. This change in recommendation is due to recent troubling trends, including an increase in the number of cancers diagnosed …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post51023/
ICARE Social Media Post May 2023
Lynch Syndrome: Skin Neoplasms
According to a recent study among individuals with Lynch Syndrome: Use the link in bio to learn more! Reference: Zhong et al. J Am Acad Dermatol. 2023;S0190-9622(23)00173-1. PMID: 36773823.
Permanent link to this article: https://inheritedcancer.net/post50623/
ICARE Social Media Post May 2023
PARP Inhibitors for Metastatic Prostate Cancer: Talazoparib
A study that compared a PARP inhibitor (talazoparib) to the standard of care (an androgen receptor inhibitor) in metastatic castration-resistant prostate cancer found that it improved progression-free survival regardless of BRCA mutation status. However, the benefit was greatest in males with a BRCA or other DNA repair gene mutation. Read the full article at this …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post50823/
ICARE Social Media Post April 2023
Cancer Risks in Older Females with a BRCA Mutation
A recent study among over 2200 females (aged 50-75) with BRCA mutations, including ICARE participants, found 379 diagnosed cancers with breast and ovarian cancer being the most common cancers observed. Overall cancer risks were 49% in BRCA1 carriers and 43% in BRCA2 carriers, and cancer risks dropped to 9% among those who had preventative removal …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post42823/
ICARE Social Media Post April 2023
Prostate Cancer in Black Men: New Genetic Variants to Explain Higher Risks
Researchers identified 9 new genetic variants that may increase the risk of developing prostate cancer. This study highlights the importance of studying diverse populations. Read the full article at this link: https://www.sciencedirect.com/science/article/abs/pii/S0302283823025617 Reference:Chen et al. Eur Urol. 2023;S0302-2838(23)02561-7. PMID: 36872133.
Permanent link to this article: https://inheritedcancer.net/post42223/
ICARE Social Media Post April 2023
New study for BRCA1, BRCA2 , or PALB2 carriers with stage 1-3 breast cancer
An open study for BRCA1, BRCA2, or PALB2 carriers with stage 1-3 breast cancer is currently recruiting! Learn more about the study by visiting https://vanderbilt.trialstoday.org/trial/NCT04584255
Permanent link to this article: https://inheritedcancer.net/post42123/
ICARE Social Media Post April 2023
PARP Inhibitors for Metastatic Prostate Cancer: Niraparib
A study that evaluated a PARP inhibitor (niraparib) in males with metastatic prostate cancer found that those with a BRCA1 or BRCA2 mutation lived longer on average than those who did not have a BRCA mutation. Read the full article at the link: https://pubmed.ncbi.nlm.nih.gov/35131040/Reference:Smith et al. Lancet Oncol. 2022;23(3):362-373. PMID: 35131040.
Permanent link to this article: https://inheritedcancer.net/post42023/
ICARE Social Media Post April 2023
Applying the Framework for Interventions to Increase Family Communication
A new study 𝗯𝘆 𝗼𝘂𝗿 𝘁𝗲𝗮𝗺 used the Framework for Developing and Evaluating Complex Interventions (FDECI) to evaluate an intervention to improve family communication of positive genetic test results for inherited cancer. The first FDECI phases were beneficial for improving the intervention and planning for continual effectiveness and future implementation phases. Use the link in …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post41023/
ICARE Newsletter Spring 2023
Community Spotlight
In 1997 when I was a junior in college, my mom called to let me know that my father had been diagnosed with prostate cancer. Luckily for him, Prostate Specific Antigen (PSA) testing had recently started which resulted in early detection and subsequent prostatectomy. Due to his diagnosis and knowing prostate cancer is an inherited …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2023span-stylecolor-56b0e4community-spotlight/
ICARE Newsletter Spring 2023
Ask the Expert
The below question was addressed by Ben Ho Park, MD, PhD, who is the Director of the Vanderbilt-Ingram Cancer Center and a Professor of Medicine in the Division of Hematology and Oncology. If you have a question you would like addressed, please email the ICARE team at ICARE@vumc.org for consideration in future newsletters. Q: As …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2023ask-the-expert/
ICARE Newsletter Spring 2023
Prostate Cancer Treatment Updates
A study to test niraparib (a PARP inhibitor) in males with metastatic prostate cancer showed that those with an inherited BRCA1 or BRCA2 (BRCA) mutation lived longer on average compared to those without a BRCA mutation. Side effects from niraparib were similar to those previously reported with PARP inhibitors.1 Another PARP inhibitor trial tested an …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2023prostate-cancer-treatment-updates/
ICARE Newsletter Spring 2023
Breast Cancer Treatment Updates
Findings from a Phase II study to evaluate the use of talazoparib (a PARP inhibitor) in individuals with advanced PALB2-mutation breast cancer showed that it appeared effective in certain patients and appeared safe (with similar adverse events as those previously reported with this drug).1 There are several Phase II trials to evaluate PARP inhibitors in …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2023-breast-cancer-treatment-updates/
ICARE Newsletter Spring 2023
Breast Cancer Screening in Male BRCA1/2 Carriers
Generally, males with breast cancer present with advanced stage disease, thought to be due to a lack of screening. While data to determine performance of breast screening through mammograms for males at inherited risk is limited, recent studies suggest that the detection rate is similar or better than for females at general population risk.1,2,3 In …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2023-breast-cancer-screening-in-male-brca1-2-carriers/
ICARE Newsletter Spring 2023
Lynch Syndrome: Colorectal Cancer Risks Revisited
While there are higher cancer risks in BRCA mutation carriers starting in the mid-20s, a recent study focused on studying cancer risks in older females aged 50-75. Of the over 2000 females in the study, which included ICARE participants, 379 cancers were found between age 50 to 75 with risks of 49% in BRCA1 carriers …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2023-lynch-syndrome-colorectal-cancer-risks-revisited/
ICARE Newsletter Spring 2023
Lynch Syndrome: Colorectal Cancer Risks Revisited
A study of 381 individuals with Lynch Syndrome in New Zealand (98 with Lynch Syndrome-associated variants in MLH1, 159 in MSH2, 103 in MSH6, and 21 in PMS2) found that risks for colorectal cancer were lower in MSH6 and PMS2 carriers, suggesting that it might be possible to spread out colonoscopy intervals for these individuals.1 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2023/
ICARE Newsletter Spring 2023
Inherited Breast Cancer: Contralateral Breast Cancer Risks
While higher risks for contralateral breast cancer (CBC) have been known for BRCA1 and BRCA2, a newly published study demonstrated that the risk of CBC is also higher for female PALB2 and CHEK2 carriers; however, no elevated risks were found for ATM carriers (Table 1).1 This information is important to study, as it may be …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2023-inherited-breast-cancer-contralateral-breast-cancer-risks/
ICARE Newsletter Spring 2023
Melanoma Risk in Li-Fraumeni Syndrome
A recently published cohort study of 483 individuals with Li-Fraumeni Syndrome (LFS) identified 113 skin cancers. The cumulative incidence of a diagnosis of any skin cancer was 36.3% by the age of 70. Specifically for melanoma, the median age at diagnosis was 42 with a 7-fold increased incidence (SIR 7.28, 95% CI 4.50-11.13) compared with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2023-melanoma-risk-in-li-fraumeni-syndrome/
ICARE Newsletter Spring 2023
National Comprehensive Cancer Network (NCCN) Guidelines Updates
Check out the full NCCN guidelines by creating a FREE account at www.nccn.org Genetic/Familial High-Risk Assessment: Breast, Ovarian, PancreaticJanuary 10th, 2023 (Version 2.2023) focused on male BRCA carriers:› Consider annual mammograms (particularly in BRCA2 carriers) starting at age 50 or 10 years before the earliest male breast cancer in the family (whichever comes first)February 13th, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2023-national-comprehensive-cancer-network-nccn-guidelines-updates/
ICARE Social Media Post March 2023
Treatment for PALB2-associated breast cancer with Talazoparib
According to a recent Phase II study, Talazoparib may benefit patients with PALB2-associated breast cancer. Findings showed that Talazoparib appeared effective and safe in certain patients with advanced disease. Read the full article at this link: https://www.nature.com/articles/s43018-022-00439-1 Reference: Gruber et al. Nat Cancer. 2022;3(10):1181-1191. PMID: 36253484.
Permanent link to this article: https://inheritedcancer.net/post32723/
ICARE Social Media Post March 2023
New Model: To identify those at high risk for Endometrial (Uterine) Cancer
Did you know endometrial cancer is the 4th most common cancer among females? Currently, clinical practice guidelines do not recommend endometrial cancer screening in the general population. A new risk prediction model was recently developed to identify those at high risk for endometrial (uterine) cancer to determine who could benefit from cancer screenings and risk-reducing …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post32623/
ICARE Social Media Post March 2023
Inherited Cancer Testing: Pheochromocytoma & Paraganglioma
Because of the high frequency of mutations found in individuals with pheochromocytoma and paraganglioma, these individuals are advised to consider genetic testing for inherited cancer.Use this link to learn more: https://pubmed.ncbi.nlm.nih.gov/35976622/ Reference: Yip, et al. JAMA Surg. 2022;157 (10):870-877. PMID: 35976622
Permanent link to this article: https://inheritedcancer.net/post32223/
ICARE Social Media Post March 2023
Metachronous Colorectal Cancer in Lynch Syndrome
Findings from a recent study: Risk factors for metachronous colorectal cancer included a history of colorectal polyps and having an MLH1 or MSH2 mutation, while protective factors included female sex and extended surgical resection. This highlights the importance of genetic testing and counseling for Lynch syndrome prior to surgery, which can influence surgical strategy and …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post31923/
ICARE Social Media Post March 2023
Colorectal Cancer Risks: Lynch Syndrome
A study on colorectal cancer risks in Lynch Syndrome patients found that:• Colorectal cancer risks were lower in MSH6 and PMS2 carriers• Colonoscopy intervals for these individuals could be longer• Incomplete polyp removal may be responsible for 1/3 of surveillance-detected colorectal cancers Read the full article with the link in bio! Reference: Lamba, et al. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post31323/
ICARE Social Media Post March 2023
New gene alert: MBD4
A new study shows germline MBD4 mutations cause an autosomal recessive tumor predisposing syndrome associated with: This highlights the importance of including MBD4 in genetic testing for polyposis and multi-tumor phenotypes to improve disease management. Use the link in bio to learn more! Reference: Palles, et al. Am J Hum Genet. 2022;109(5):953-960. PMID: 35460607.
Permanent link to this article: https://inheritedcancer.net/post31223/
ICARE Social Media Post March 2023
BGREAT December 2022 Newsletter
Check out the latest edition of our B-GREAT newsletter for updates about inherited cancers in the context of racial inequalities in healthcare. You can read the newsletter by visiting 👇https://bgreatinitiative.inheritedcancer.net/wp-content/uploads/BGREAT-December-2022-Newsletter.pdf Please feel free to share with family members, friends, and/or your healthcare providers.
Permanent link to this article: https://inheritedcancer.net/post30623/
ICARE Social Media Post March 2023
Melanoma in TP53 /Li-Fraumeni Syndrome
A new study found a 7-fold increased incidence of melanoma in individuals with TP53/Li-Fraumeni Syndrome compared with the general population:• Average age was 42• Risk to age 40 was 2.8%• Risk to age 60 was 7.8%• Risk to age 70 was 14.9% Learn more at the link below!https://pubmed.ncbi.nlm.nih.gov/35183552/ Reference: Hatton, et al. J Invest Dermatol. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post30723/
ICARE Social Media Post February 2023
Why is learning about inherited cancer important to guide care?
Why have genetic testing? Genetic testing for inherited cancer can help guide care, including:• Cancer treatment plans, such as chemotherapy, drugs, surgery, and radiation• Cancer screening and prevention Sharing genetic test results can also help family members understand their cancer risks.
Permanent link to this article: https://inheritedcancer.net/post22423/
ICARE Social Media Post February 2023
ATM Gene and Breast Cancer Risk for: c.7570G>C Mutation
While ATM mutations are generally associated with a 25-30% lifetime risk for breast cancer, emerging information suggests that the risk in those with a c.7570G>C mutation may be higher. Once confirmed, this information may be important to guide cancer screening and risk-reducing surgery decisions. Use this link to learn more: https://onlinelibrary.wiley.com/doi/10.1002/ijc.34305
Permanent link to this article: https://inheritedcancer.net/post40623/
ICARE Newsletter Spring 2022
Reducing Hereditary Cancer Act of 2021
On February 16th, 2022, the Reducing Hereditary Cancer Act (S.3656) was introduced to the U.S. Senate with the goal of ensuring Medicare beneficiaries have coverage for potentially life-saving inherited cancer genetic counseling and testing as well as recommended screening and risk-reducing procedures, when medically necessary and appropriate. Currently, Medicare only covers genetic testing for beneficiaries …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-reducing-hereditary-cancer-act-of-2021/
ICARE Newsletter Spring 2022
Community Spotlight
Genetic testing sounded like a futuristic concept when it originally came to our attention. It sounded something to the likes of cloning. We didn’t really understand what it was and why it was important, but rather thought it sounded like something out of science fiction. Thankfully, our medical providers and genetic counselors stuck with us …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-community-spotlight/
ICARE Newsletter Spring 2022
Ask the Expert
The below question was addressed by ICARE Founder, Dr. Tuya Pal, and her oncology colleague, Dr. Sonya Reid. Dr. Pal is a Professor of Genetic Medicine, Ingram Professor of Cancer Research, and the Associate Director for Cancer Health Disparities at VanderbiltIngram Cancer Center. Dr. Reid is an Assistant Professor of Hematology/Oncology at Vanderbilt University Medical …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-ask-the-expert/
ICARE Newsletter Spring 2022
Inherited Cancer Treatment Updates
Lynch Syndrome Carriers with Advanced Uterine Cancer: Treatment with PembrolizumabWomen with Lynch Syndrome are at high risk for uterine cancer. The type of uterine cancer they develop has the tumorcharacteristic of being ‘MSI-H’. A new study indicated treatment with pembrolizumab (Keytruda) resulted in benefit inpatients with MSI-H advanced uterine cancer. Von Hippel-Lindau Patients: Treatment of …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-inherited-cancer-treatment-updates/
ICARE Newsletter Spring 2022
BRCA1/2 Oral Contraceptives and Breast Cancer
A new study found that among BRCA1/2 carriers, oral contraceptive use strongly lowered cancer risk over one’s lifetime, even though at first, they raise risks of breast, ovarian, and endometrial cancer.Schrijver et al. J Natl Cancer Inst. 2022 Jan. PMID: 35048954. Social media post February 15th, 2022. Available at:https://tinyurl.com/post21522.
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-brca1-2-oral-contraceptives-and-breast-cancer/
ICARE Newsletter Spring 2022
BRCA1/2 and Male Cancer Risks
A recent international study found that male BRCA1 and BRCA2 carriers have a higher risk for breast, pancreatic, and stomach cancer. Additionally, male BRCA2 carriers were found to have higher risks for prostate cancer. See the below table for the specific risk levels: Li et al. J Clin Oncol. 2022 Jan. PMID: 35077220. Social media …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-brca1-2-and-male-cancer-risks/
ICARE Newsletter Spring 2022
Lynch Syndrome and Prostate Cancer
A new study suggests men with certain MSH2 and MSH6 mutations have higher risks of prostate cancer and may be candidates for PSA screening. Bancroft et al. Lancet Oncol. 2021 Nov. PMID: 34678156. Social media postDecember 17th, 2021. Available at: https://tinyurl.com/post121721
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-lynch-syndrome-and-prostate-cancer/
ICARE Newsletter Spring 2022
Polygenic Risk Scores and Inherited Breast Cancer Genes: BRCA1/2, PALB2, CHEK2, and ATM
Breast MRIs are advised in women with >20% lifetime risk of breast cancer. A new study showed that breast cancer risks in BRCA1, BRCA2 and PALB2 carriers remained higher than 20%, regardless of whether polygenic risk scores (PRS) were done, suggesting this is of limited help in refining screening. In contrast, PRS downgraded breast cancer …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-polygenic-risk-scores-and-inherited-breast-cancer-genes-brca1-2-palb2-chek2-and-atm/
ICARE Newsletter Spring 2022
Li-Fraumeni Syndrome and Cancer Risks
A new study reported on differences in the TP53 mutations between patients who met vs those who did not meet Li-Fraumeni Syndrome (LFS) testing criteria. Several variants were identified multiple times in those who did and did not meet LFS clinical criteria: p.R175, p.G245, p.R248, p.R273, and p.R282. Other variants were exclusively found in those …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-li-fraumeni-syndrome-and-cancer-risks/
ICARE Newsletter Spring 2022
CDH1 and Stomach Cancer
A recent study of women with hereditary lobular breast cancer due to CDH1 mutations found high rates ofundetected stomach cancer (with signet ring features). The findings suggested that among CDH1 carriers, there werevery high risks for stomach cancer, even when family history of stomach cancer was absent.Gamble et al. JAMA Surg. 2021 Oct. PMID: 34643667. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-cdh1-and-stomach-cancer/
ICARE Newsletter Spring 2022
ATM and Pancreatic Cancer
A new article reported that pancreatic cancer risks are morethan 6-fold higher in individuals with an ATM mutation.Pancreatic cancer risks by age were:› 1.1% by age 50› 6.3% by age 70› 9.5% by age 80Hsu, et al. JAMA Oncol. 2021 Sep. PMID: 34529012. Social media post October 1st, 2021. Available at: https://tinyurl.com/post100121.
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-spring-2022-atm-and-pancreatic-cancer/
ICARE Newsletter Fall 2022
Community Spotlight
When I was just 8 years old my mother was diagnosed with a very aggressive breast cancer. I didn’t reallyunderstand the concept of cancer at that age, but I knew what was happening was terrible. After manysurgeries and treatments, she passed away 2 years later at the age of 35. There was no hereditary cancertesting …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2022-community-spotlight/
ICARE Newsletter Fall 2022
Ask the Expert
The below question was addressed by Dr. Kamran Idrees, Chief of the Division of Surgical Oncology & Endocrine Surgery, Associate Professor of Surgery, Ingram Associate Professor of Cancer Research, and Director of Pancreatic and Gastro Intestinal Surgical Oncology at Vanderbilt-Ingram Cancer Center. Dr. Idrees’ research has focused on colorectal cancer, liver metastases, and pancreatic cancer …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2022-ask-the-expert/
ICARE Newsletter Fall 2022
ATM -associated Cancer Risks
Female ATM carriers have approximately a 2-fold risk, on average, for developing breast cancer, which was confirmed through two large studies that reported risks (odds ratio) as 2.10 (95% CI, 1.71–2.57)1 and 1.82 (95% CI, 1.46–2.27). Both studies reported an association with ER+ tumors. Another recent study estimated breast cancer risks to be 13% by …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2022-atm-associated-cancer-risks/
ICARE Newsletter Fall 2022
Screening & Treatment Updates: New Modeling Analysis About Breast Cancer Screening in ATM and CHEK2 Carriers
Using information from twelve prior population-based studies, a modeling analysis was done to look at when to start mammography and breast MRI in females with inherited mutations in genes including ATM and CHEK2. Overall, findings showed that starting annual MRI screening between age 30 to 35 and mammography at age 40 may lower death from …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2022-screening-treatment-updates-new-modeling-analysis-about-breast-cancer-screening-in-atm-and-chek2-carriers/
ICARE Newsletter Fall 2022
Screening & Treatment Updates: Pancreatic Cancer
A recent small study suggests that immunotherapy may benefit patients with refractory pancreatic or biliary cancer who have inherited a mutation in the homologous recombination deficiency (HRD) genes, BRCA1, BRCA2, and RAD51C.Another new study reported that in BRCA1/2 carriers with pancreatic cancer, maintenance treatment with Olaparib may be of benefit. Findings showed that with Olaparib, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2022-screening-treatment-updates-pancreatic-cancer/
ICARE Newsletter Fall 2022
Which Genes Are Confirmed as ‘Inherited Breast Cancer Genes’?
There were two large studies published early last year that evaluated which genes raise risks for breast cancer, including breast cancer patients from many centers worldwide, representing the largest available datasets to look at this question. These efforts were led by the worldwide Breast Cancer Association Consortium (BCAC)1 and the United States-based CARRIERS consortium. The …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2022-which-genes-are-confirmed-as-inherited-breast-cancer-genes/
ICARE Newsletter Fall 2022
Inherited Cancer Genes: New Associations
A new study led by colleagues at Vanderbilt University Medical Center, including our clinical geneticist colleague, Dr. Georgia Wiesner, evaluated 23 hereditary cancer genes and found 19 new gene associations including 7 new associations with cancer and 12 new associations with noncancer diseases. The associations with cancer versus other conditions is included in the table. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2022-inherited-cancer-genes-new-associations/
ICARE Newsletter Fall 2022
BRCA1/2 Cancer Risk Updates
During preventive surgery to remove the ovaries and fallopian tubes (called a risk-reducing salpingo-oophorectomy orRRSO), a new study found that the detection of tubal intraepithelial carcinoma predicts the risk of later peritonealcancer.1 These findings show the importance of timely RRSO and the need to do a careful pathology exam of the ovaries and fallopian tubes …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2022-brca1-2-cancer-risk-updates/
ICARE Newsletter Fall 2022
National Comprehensive Cancer Network (NCCN) Guidelines Updates
Check out the full NCCN guidelines by creating a FREE account at www.nccn.org Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic – Released September 7th, 2022› Testing eligibility based on personal history of any type of breast cancer in females was updated from age ≤45 to ≤50 making more females with breast cancer eligible for testing …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2022-national-comprehensive-cancer-network-nccn-guidelines-updates/
ICARE Newsletter Fall 2021
Resource Spotlight: National Cancer Institute’s Cancer Genetics PDQ
Looking for a place to get the latest curated information about inherited cancers? Look no further… Provide evidence-basedsummaries about inheritedcancer genetics for healthprofessionals and patients Summaries updated basedon monthly review ofpublished literature Written and maintained by amultidisciplinary Editorial Board ofexperts in medical genetics, oncology,and related specialties Visit https://tinyurl.com/NCIPDQ and https://www.cancer.gov/publications/pdq/editorial-boards/genetics for more information
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2021-resource-spotlight-national-cancer-institutes-cancer-genetics-pdq/
ICARE Newsletter Fall 2021
Direct-to-Consumer Tests: Not Reliable to Detect BRCA1/2 Mutations
Researchers reported that single nucleotide polymorphism (SNP) tests, which are used by DTC tests such as 23andMe, are not reliable in identifying the majority of BRCA1/2 mutations or other inherited cancer gene mutations. The SNP test used by many DTC ancestry and DNA companies is designed to detect common traits many people share. However, when …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2021-direct-to-consumer-tests-not-reliable-to-detect-brca1-2-mutations/
ICARE Newsletter Fall 2021
Reducing Hereditary Cancer Act of 2021
Under current Medicare guidelines, only those with “signs, symptoms, complaints, or personal histories of disease” meet the criteria for medical services coverage. Thus, genetic testing is only covered for those already diagnosed with cancer, regardless of family history. If someone without cancer has an inherited mutation that increases cancer risk (e.g., BRCA1/2), Medicare may not …
Continue reading
Permanent link to this article: https://inheritedcancer.net/div-classboxedspan-stylecolor-whiteh6icare-newsletter-fall-2021-h6-span-divbrcenterh4span-stylecolor-56b0e4-reducing-heredit/
ICARE Newsletter Fall 2021
Community Spotlight
At the age of 51, my first and only colonoscopy revealed 100 polyps in my colon, rectum, and anuseven though I had no symptoms or family history. I was immediately referred to a Certified GeneticCounselor at Tripler Army Medical Center in Hawai’i. Germline DNA testing revealed I had attenuatedfamilial adenomatous polyposis (AFAP), due to an …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2021-community-spotlight/
ICARE Newsletter Fall 2021
Ask the Expert
In each newsletter, we give participants the opportunity to have their questions addressed by experts in thefield. This question was addressed by Kerry Schaffer, MD, medical oncologist at Vanderbilt University MedicalCenter with a focus on urological cancers. Q. Is there enough information to consider using PARP inhibitors to treat inherited forms ofprostate cancer? A. Over …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2021-ask-the-expert/
ICARE Newsletter Fall 2021
>
Polygenic Risk Scores and Breast Cancer Risks: BRCA1/2, PALB2, CHEK2, ATM , and beyond!
A recent study found use of a polygenic risk score (PRS) modified the estimated riskof breast cancer among both carriers and non-carriers of inherited breast cancerpredisposition genes. Taking PRS into account, more than 95% of BRCA1, BRCA2,and PALB2 carriers had greater than 20% lifetime risks of breast cancer. In contrast,among ATM and CHEK2 carriers without …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-fall-2021-polygenic-risk-scores-and-breast-cancer-risks-brca1-2-palb2-chek2-atm-and-beyond/
ICARE Social Media Post February 2023
Risk for second breast cancer in premenopausal female BRCA1, BRCA2, PALB2 and CHEK2 carriers
Did you know female BRCA1, BRCA2, CHEK2, or PALB2 carriers have much higher risks for contralateral breast cancer?This information is important to guide cancer screening and risk reduction strategies. Use the link in bio to learn more!
Permanent link to this article: https://inheritedcancer.net/post40123/
Newsletter Fall 2021
Inherited Cancer Treatment Updates
Early-stage, high-risk breast cancer in BRCA carriers: Results of the highly awaited phase 3 OlympiA trial showed promising results for EARLY STAGE (i.e., localized Stage 2-3) high-risk breast cancer patients with a BRCA mutation who were treated with a PARP inhibitor (olaparib) in the adjuvant setting (i.e., AFTER surgery).1 Early-stage breast cancer in this trial …
Continue reading
Permanent link to this article: https://inheritedcancer.net/newsletter-fall-2021-inherited-cancer-treatment-updates/
Newsletter Fall 2021
Modifying Risks in BRCA Carriers
Breast cancer risks: A risk-reducing salpingo-oophorectomy (i.e., removal of both ovaries and fallopian tubes) in BRCA carriers was associated with a reduced risk of breast cancer within five years after surgery, with evidence of longer-term risk reduction among those with BRCA1 variants.1 Ovarian cancer risks: A new study reported that the use of oral contraceptives …
Continue reading
Permanent link to this article: https://inheritedcancer.net/newsletter-fall-2021-modifying-risks-in-brca-carriers/
Newsletter Fall 2021
Breast Cancer Risks Remain High in PALB2 & BRCA
A new study found that lifetime breast cancer risk is 15% or more in female BRCA1, BRCA2, and PALB2 carriers over age 65. This level of risk warrants consideration for continuing breast MRI.1 These results are similar to those of a study that included ICARE participants,2 which reported the risk of developing breast cancer remains …
Continue reading
Permanent link to this article: https://inheritedcancer.net/newsletter-fall-2021-breast-cancer-risks-remain-high-in-palb2-brca/
Newsletter Fall 2021
Prostate Cancer and PALB2
A new Polish study based on two specific founder mutations in PALB2 reported that mutations in this gene may predispose to an aggressive, lethal form of prostate cancer.1 The investigators studied PALB2 prostate cancer risks, characteristics, and outcomes in almost 5,500 men with prostate cancer and compared them to over 8,000 cancer-free adults from Poland. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/newsletter-fall-2021-prostate-cancer-and-palb2/
Newsletter Fall 2021
PALB2 : Increasingly Recognized as the Third Most Important Inherited Breast Cancer Gene
In May 2021, a clinical practice resource was released by the American College of Medical Genetics and Genomics (ACMG) from a global team of cancer genetics specialists (see figure) to help guide the care of PALB2 carriers.1 PALB2 is considered the third most important breast cancer risk gene, after BRCA1 and BRCA2, with PALB2 carriers …
Continue reading
Permanent link to this article: https://inheritedcancer.net/div-classboxedspan-stylecolor-whiteh6newsletter-fall-2021-h6-span-divbrcenterh4span-stylecolor-56b0e4-ipalb2-i-increas/
Newsletter Fall 2021
Updates to NCCN Genetic/Familial High-Risk Assessment
Breast, Ovarian, and Pancreatic Guidelines V.1.2022: Released August 11th, 2021 Colorectal Cancer Guidelines V.1.2021: Released May 11th, 2021 Check out the full NCCN guidelines by creating a FREE account at www.nccn.org
Permanent link to this article: https://inheritedcancer.net/newsletter-fall-2021-updates-to-nccn-genetic-familial-high-risk-assessment/
ICARE Social Media Post January 2023
Inherited Breast Cancers Across Populations
Did you know that BRCA1/2 are amongst the most well-studied genes, yet most BRCA1/2 studies have been done in White populations? This means our knowledge about genes and risks comes primarily from White populations. • Some research suggests that BRCA1/2 gene mutations may be more common in young Black women with breast cancer. • Even …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12423-2/
ICARE Social Media Post January 2023
Updates to NCCN Guidelines: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic
The National Comprehensive Cancer Network (NCCN) just released updated breast, ovarian, and pancreatic cancer guidelines which included updated breast cancer screening recommendations for male BRCA carriers (particularly male BRCA2 carriers). It is now recommended that male BRCA carriers consider annual mammograms starting at age 50 or 10 years before the earliest male breast cancer in …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12423/
ICARE Social Media Post January 2023
New Contralateral Breast Cancer Risk Prediction Model for BRCA1/2 Carriers
A new risk prediction model was developed to assess the risk of contralateral breast cancer in BRCA1/2 carriers. Risks are higher with:• Younger age at first breast cancer• Close family member with breast and/or ovarian cancer• Mutation located near the 3′ region of the gene Risks are lower with:• Endocrine therapy Use this link in …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11423/
ICARE Social Media Post January 2023
Why is diversity in genomics studies important?
Did you know that about 80% of genomics data comes from European populations, yet they only make up about 16% of the world population? This bias means Europeans stand to benefit the most, while important associations for other ancestry groups may be missed. Not including diverse populations in genomics research can further WIDEN disparities!
Permanent link to this article: https://inheritedcancer.net/post10823/
ICARE Social Media Post December 2022
Negative or Uninformative Test Results
A new study demonstrates the importance of retesting patients with previously uninformative cancer genetics evaluations, especially in high risk individuals. Read the full article at this link for more info: https://pubmed.ncbi.nlm.nih.gov/34545504/ 🔗 Reference: Dettwyler et al. Fam Cancer. 2022; DOI: 10.1007/s10689-021-00276-8. PMID: 34545504
Permanent link to this article: https://inheritedcancer.net/post121322/
ICARE Social Media Post December 2022
Bilateral Mastectomy in BRCA1/2, PALB2, ATM, & CHEK2 Carriers
A recent study including data from ICARE participants found similar rates of bilateral mastectomy across high (BRCA1, BRCA2, PALB2) and moderate (ATM, CHEK2) penetrance genes. The high rates of bilateral mastectomies seen in those with moderate penetrance genes is concerning for overtreatment. Use the link to learn more: https://jamanetwork.com/journals/jamaoncology/fullarticle/2797978?guestAccessKey=fe9a3a20-8623-4feb-a0c5-315ad43a8fcb&utm_source=jps&utm_medium=email&utm_campaign=author_alert-jamanetwork&utm_content=author-author_engagement&utm_term=1m Reference: Reid et al. Receipt of …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post120822/
ICARE Social Media Post December 2022
Cancer Screen Week
This week is Cancer Screen Week! This week we would like to highlight the importance of screening for inherited cancer to help prevent cancer and find it early. Talk to your healthcare provider today about what screenings are best for you.
Permanent link to this article: https://inheritedcancer.net/post120622/
ICARE Social Media Post November 2022
Cancer Risks: CHEK2
A newly published study reports that the type of CHEK2 mutation leads to different cancer risks. Read the full article at this link: https://jamanetwork.com/journals/jamaoncology/fullarticle/2796767 Reference: Bychkovsky BL, et al. JAMA Oncol. 2022 Sep 22;e224071. PMID: 36136322.
Permanent link to this article: https://inheritedcancer.net/post112122-2/
ICARE Social Media Post November 2022
Fall 2022 Ask the Expert
In every ICARE newsletter we give our participants the opportunity to have a question addressed by an expert in the field. In the latest edition, Dr. Kamran Idrees, Chief of the Division of Surgical Oncology & Endocrine Surgery, Associate Professor of Surgery, Ingram Associate Professor of Cancer Research, and Director of Pancreatic and Gastro-Intestinal Surgical …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post111122/
ICARE Social Media Post November 2022
Guidelines for Hereditary Breast Cancer Treatment
The Expert Panel Consensus, chaired by the Society of Surgical Oncology with the American Society of Clinical Oncology and the American Society of Radiation Oncology, published new recommendations for hereditary breast cancer treatment. Learn more at https://pubmed.ncbi.nlm.nih.gov/32898012/ Reference: Corso & Magnoni. Eur J Cancer Prev. 2021;30(4):311-314. PMID: 32898012.
Permanent link to this article: https://inheritedcancer.net/post110822/
ICARE Social Media Post November 2022
ICARE Fall 2022 Newsletter
The ICARE Fall 2022 Newsletter is now available! Check out this latest edition for recent research and clinical updates, a Q&A with Dr. Kamran Idrees from Vanderbilt-Ingram Cancer Center, and an inspiring community spotlight piece. You can read and download a copy of the newsletter at: https://inheritedcancer.net/newsletters/ Please feel free to share with family members, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post110422/
ICARE Social Media Post October 2022
Tool for Inherited Cancer Predisposition Counseling and Testing Study (TIPS)
Have you already had genetic testing for inherited cancer and want to get more education and information about what your results mean for you? We provide free education about inherited cancer risk through our Tool for Inherited Cancer Predisposition Counseling and Testing Study (TIPS). Visit https://redcap.link/TIPS and complete our survey. Once completed, we provide:• an …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102422/
ICARE Social Media Post October 2022
October 21 – National Mammography Day
National Mammography Day is observed annually on the third Friday of every October. Today acts as reminder to everyone that early detection is the best defense against breast cancer!
Permanent link to this article: https://inheritedcancer.net/post102122/
ICARE Social Media Post October 2022
October 13 Metastatic Breast Cancer Awareness Day
October 13th is Metastatic Breast Cancer Awareness Day. Today is a day to raise awareness about the importance of furthering cancer research.
Permanent link to this article: https://inheritedcancer.net/post101322/
ICARE Social Media Post October 2022
NCCN Familial/Genetic High Risk Assessment: Breast, Ovarian & Pancreatic Guidelines single post 3
On September 7th, 2022, the National Comprehensive Cancer Network (NCCN) released new breast, ovarian, and pancreatic cancer guidelines. In these new guidelines, genetic testing eligibility based on age at breast cancer diagnosis in females was updated from ≤ 45 to ≤ 50 making more females with breast cancer eligible for testing regardless of family history …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post101022/
ICARE Social Media Post October 2022
CanRisk Tool
Have you heard of the CanRisk Tool?The CanRisk Tool provides health care professionals a user-friendly resource to carry out breast and ovarian cancer risk predictions. It is the first freely accessible cancer risk prediction program!Read the full article at the link: https://pubmed.ncbi.nlm.nih.gov/33335023/ Reference: Carver et al. Cancer Epidemiol Biomarkers Prev. 2021 Mar;30(3):469-473. PMID: 33335023.
Permanent link to this article: https://inheritedcancer.net/post100722-2/
ICARE Social Media Post October 2022
NCCN Familial/Genetic High Risk Assessment: Breast, Ovarian & Pancreatic Guidelines – single post 2
On September 7th, 2022, the National Comprehensive Cancer Network (NCCN) released new breast, ovarian, and pancreatic cancer guidelines. In these new guidelines, the age to start breast MRI screening in female ATM and CHEK2 carriers was updated from age 40 to 30-35 making more females eligible for screening at an earlier age.Check out the new …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post100222/
ICARE Social Media Post October 2022
Ovarian Cancer Risks: Weight Gain in BRCA1/2 Carriers
A study found that adult weight gain is a risk factor for ovarian cancer. This highlights the importance for BRCA1/2 carriers to maintain a healthy weight throughout adulthood.Read the full article to learn more! https://pubmed.ncbi.nlm.nih.gov/34426412/Reference: Kim et al. Cancer Epidemiol Biomarkers Prev. 2021 Nov;30(11):2038-2043. PMID: 34426412.
Permanent link to this article: https://inheritedcancer.net/post100122/
ICARE Social Media Post September 2022
Breast cancer risks: BRCA1/2 carriers with risk reducing salpingo-oophorectomy (RRSO)
Three recent studies found varying results when evaluating breast cancer risk in BRCA1/2 carriers with risk-reducing salpingo-oophorectomy (RRSO). Use these links to learn more: https://pubmed.ncbi.nlm.nih.gov/31948486/ https://pubmed.ncbi.nlm.nih.gov/33630024/ https://pubmed.ncbi.nlm.nih.gov/35216860/
Permanent link to this article: https://inheritedcancer.net/post92722/
ICARE Social Media Post September 2022
Hormonal Replacement Therapy (HRT) in BRCA1 and BRCA2 carriers
A recent study showed hormone replacement therapy (HRT) is reasonable to offer to BRCA1 and BRCA2 carriers who underwent risk-reducing salpingo-oophorectomy (RSSO). Read the full article at this link: https://pubmed.ncbi.nlm.nih.gov/32143914/Reference: Mills, et al. Gynecol Oncol; 2020 Jun;157(3):706-710. PMID: 32143914.
Permanent link to this article: https://inheritedcancer.net/post2922/
ICARE Social Media Post September 2022
Bilateral Oophorectomy: Breast Cancer Risks in BRCA1 Carriers
A recent study, including ICARE participants, found preventive removal of the ovaries does not reduce breast cancer risk in BRCA1 carriers. These findings highlight that study bias can influence results.Read the full article at this link: https://pubmed.ncbi.nlm.nih.gov/35477169/Reference: Kotsopoulos et al. Cancer Epidemiol Biomarkers Prev. 2022 Jul 1;31(7):1351-1358. PMID: 35477169.
Permanent link to this article: https://inheritedcancer.net/icare-social-media-post-september-2022-bilateral-oophorectomy-breast-cancer-risks-in-brca1-carriers/
ICARE Social Media Post September 2022
NCCN Familial/Genetic High Risk Assessment: Breast, Ovarian & Pancreatic Guidelines – single post 1
On September 7th, 2022, the National Comprehensive Cancer Network (NCCN) released new breast, ovarian, and pancreatic cancer guidelines. In this new version, a table was added at the end of the guidelines to summarize inherited cancer content across 𝗮𝗹𝗹 NCCN guidelines, including live links to the guidelines and sections referenced (see page 54 on the …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post92222/
ICARE Social Media Post September 2022
/span>
Risk of Ovarian Cancer In BRCA1/2 Carriers: Oral Contraceptives and Implants
A new study suggests oral contraceptives and implants lower risk of ovarian cancer in BRCA1/2 carriers.Read the full article at the link: https://pubmed.ncbi.nlm.nih.gov/35063280/Reference: Xia et al. Gynecol Oncol. 2022;164(3):514-521. doi:10.1016/j.ygyno.2022.01.014. PMID: 35063280.
Permanent link to this article: https://inheritedcancer.net/post92122/
ICARE Social Media Post September 2022
ATM: Breast Cancer Risks
A recent study found that even though ATM has been clearly identified as a breast cancer risk gene with an estimated risk of 13% up to age 80, variant-specific penetrance estimates are needed to guide risk management plans for those with pathogenic mutations.Read the full article at the link: https://pubmed.ncbi.nlm.nih.gov/35365198/Reference: Renault et al. Breast Cancer …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post91922/
ICARE Social Media Post September 2022
NCCN Familial/Genetic High Risk Assessment: Breast, Ovarian & Pancreatic Guidelines
On September 7th, 2022, the National Comprehensive Cancer Network (NCCN) released new breast, ovarian, and pancreatic cancer guidelines.Check out the new guidelines at:https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf
Permanent link to this article: https://inheritedcancer.net/post91222/
ICARE Social Media Post September 2022
September ICARE Genetics Case Conference
We are excited to announce that our next ICARE genetics case conference, focused on TERT mutations in myeloid malignancies, will be broadly accessible. We are honored to have Mary Armanios, MD as our guest expert for this conference, in which she will provide a featured presentation and case commentary. We encourage you to join us …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post90822/
ICARE Social Media Post August 2022
Nipple sparing mastectomy: BRCA1/2 carriers
According to a recent study, a bilateral prophylactic nipple-sparing mastectomy appears to be at least equally as safe as other types of mastectomy for preventing breast cancer. Read the full article at the link: https://pubmed.ncbi.nlm.nih.gov/34342702/Reference: Stanek et al. Aesthetic Plast Surg. 2022 Apr;46(2):706-711. PMID: 34342702.
Permanent link to this article: https://inheritedcancer.net/post82922/
ICARE Social Media Post August 2022
ATM: Breast Cancer Risks
ATM variants are linked to an increased risk of breast cancer, with the ATM V2424G variant causing the highest risks and ATM D1853V, L546V, and S707P variants being the least predictive.Read the full article here: https://pubmed.ncbi.nlm.nih.gov/33402103/Reference: Moslemi, et al. BMC Cancer. 2021 Jan 5;21(1):27. PMID: 33402103.
Permanent link to this article: https://inheritedcancer.net/post81922/
ICARE Social Media Post August 2022
Ovarian Cancer Risks: Aspirin
A new report found that regular aspirin use was associated with a 13% reduction in ovarian cancer risk. Read the full article at the link: https://ascopubs.org/doi/abs/10.1200/JCO.21.01900Reference: Hurwitz et al. J Clin Oncol. 2022. PMID: 35867953.
Permanent link to this article: https://inheritedcancer.net/post81222/
ICARE Social Media Post August 2022
Pancreatic Cancer Treatment
A new study reports that maintenance treatment with Olaparib may benefit BRCA1/2 carriers with pancreatic cancer. These findings demonstrated:long-term survival was more commontime to subsequent therapy was prolongedRead the full article at the link: https://ascopubs.org/doi/pdf/10.1200/JCO.21.01604Reference: Kindler et al. J Clin Oncol. 2022; JCO2101604. PMID: 35834777.
Permanent link to this article: https://inheritedcancer.net/post80522/
ICARE Social Media Post July 2022
Pancreatic Cancer Screening
A recent study found that earlier diagnosis improved survival in people at high risk of pancreatic cancer.High risk was defined based on:family history and/orinherited gene mutation (BRCA1, BRCA2, CDKN2A, Lynch Syndrome genes, PALB2, ATM, and STK11)Read the article at the link: https://ascopost.com/news/june-2022/outcomes-of-pancreas-surveillance-in-the-caps5-study-and-total-caps-cohort/Reference: Dbouk, et al. J Clin Oncol. 2022 Jun 15:JCO2200298. doi: 10.1200/JCO.22.00298. PMID: 35704792.
Permanent link to this article: https://inheritedcancer.net/post72622/
ICARE Social Media Post June 2022
ATM: High and Moderate Risks for Multiple Cancers
A recent study found that pathogenic variants in the Ataxia Telangiectasia Mutated (ATM) gene are associated with multiple cancers. Specifically, moderate-to-high risks for pancreatic, prostate, gastric, and invasive ductal breast cancers, and low-to-moderate risks for ductal carcinoma in situ, male breast cancer, ovarian cancer, colorectal cancer, and melanoma.This provides more data to guide risks and …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post62122/
ICARE Social Media Post June 2022
Refractory Pancreatic or Biliary Cancer
A recent small study suggests that immunotherapy may be beneficial for patients with refractory pancreatic or biliary cancer who have inherited homologous recombination deficiency (HRD) genes, BRCA1, BRCA2, and RAD51C.Check out the full article to learn more at 👇https://jamanetwork.com/journals/jamaoncology/article-abstract/2791557Reference: Terrero et al. JAMA Oncol. 2022 Apr:e220611. doi:10.1001/jamaoncol.2022.0611. PMID: 35446342.
Permanent link to this article: https://inheritedcancer.net/post61322/
ICARE Social Media Post June 2022
Maternal Malignancies May Be Identified Through Noninvasive Prenatal Test Results
A new study found that results of noninvasive prenatal testing for fetal aneuploidy screening using cell-free DNA derived from maternal plasma raised suspicion of maternal malignancy in a small proportion of pregnant women. However, among those with suspicious findings, the risk of a confirmed malignancy was quite high.Check out the full article athttps://ascopubs.org/doi/full/10.1200/JCO.21.02260Reference: Heesterbeek et …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post60822/
ICARE Social Media Post June 2022
New Variants Linked to Hereditary Cancer
A new study evaluated 23 hereditary cancer genes and found 19 new gene associations, including 7 new associations with cancer and 12 new associations with non-neoplastic diseases. Specifically, the below genes were found to have an increased risk of disease:APC: benign liver/bile duct tumors, gastritis, and duodenitisATM: stomach cancer and pancreatic cancerBRCA1/2: ovarian cystsCHEK2: leukemia …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post60122/
ICARE Social Media Post May 2022
BRCA Carriers with Risk-Reducing Salpingo-oophorectomy: Risk of Peritoneal Carcinomatosis
A new study found that among BRCA1/2 carriers, the presence of tubal intraepithelial carcinoma during risk-reducing salpingo-oophorectomy (i.e., preventive surgery to remove the ovaries and fallopian tubes) predicts the risk of later peritoneal cancer. These findings demonstrate:the importance of timely risk-reducing removal of the ovaries and fallopian tubesthat it is VERY important to have a …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post52422/
ICARE Social Media Post May 2022
Breast Cancer Genes in Women of African Ancestry
A recent study in women of African ancestry confirmed genes previously identified to have associations with breast cancer risk (BRCA1, BRCA2, PALB2, ATM, TP53, NF1, and CHEK2) and provided new evidence of breast cancer risk for RAD51C and RAD51D, which was identified previously in European ancestry populations.Check out the full article at 👇https://pubmed.ncbi.nlm.nih.gov/35396981/Reference: Díaz-Zabala, et …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post51722/
ICARE Social Media Post May 2022
Melanoma Treatment Update
Results from a small study suggest that melanomas in CDKN2A carriers may have better response rates to immunotherapy compared to non-carriers.Read the article to learn more!https://pubmed.ncbi.nlm.nih.gov/30291219/Reference: Helgadottir et al. J Med Genet. 2020 May;57(5): 316-321. PMID: 30291219.
Permanent link to this article: https://inheritedcancer.net/post51022/
ICARE Social Media Post May 2022
CDKN2A and Melanoma
Did you know that CDKN2A mutations are found in 35-43% of families with three or more family members with melanoma?Read the article to learn more!https://pubmed.ncbi.nlm.nih.gov/33945383/Reference: Pissa et al. Acta Oncol. 2021 Jul;60(7):888-896. PMID: 33945383.
Permanent link to this article: https://inheritedcancer.net/post50622/
ICARE Social Media Post April 2022
Spring 2022 Ask the Expert
In every ICARE newsletter we give our participants the opportunity to have a question addressed by an expert in the field. In the latest edition, ICARE Founder, Dr. Tuya Pal, and her oncology colleague, Dr. Sonya Reid, discuss the types of inherited breast cancer and whether having an inherited form of breast cancer affects survival.Check …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post42922/
ICARE Social Media Post April 2022
Disparities: PARP Inhibitors & Ovarian Cancer
A recent study found that non-Hispanic Black and Hispanic women are underrepresented in clinical trials evaluating PARP inhibitor use among ovarian cancer patients; therefore, these trials do not accurately represent the ovarian cancer patient population. These findings indicate initiatives to increase diversity in clinical trials are needed. Read the full article for more information 👇https://www.gynecologiconcology-online.net/article/S0090-8258(22)00071-3/fulltext#secst0030Reference: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post42222/
ICARE Social Media Post April 2022
BRCA1/2 Carriers with Breast Cancer: Olaparib & Survival
A new study found that adjuvant olaparib significantly extended survival in BRCA1/2 carriers with HER2-negative high-risk early-stage breast cancer. Learn more at the following link: https://www.healio.com/news/hematology-oncology/20220323/adjuvant-olaparib-prolongs-survival-for-certain-patients-with-early-breast-cancer
Permanent link to this article: https://inheritedcancer.net/post41922/
ICARE Social Media Post April 2022
Metastatic Prostate Cancer & Olaparib
Read the full article for more information!https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(22)00017-1/fulltextReference: Thiery-Vuillemin, et al. Lancet Oncol. 2022 Mar;23(3):393-405. PMID: 35157830.
Permanent link to this article: https://inheritedcancer.net/post40822/
Permanent link to this article: https://inheritedcancer.net/post40522/
ICARE Social Media Post April 2022
Breast Cancer Mortality & MRI Screening: ATM, CHEK2, and PALB2
A new study suggests that annual breast MRI screening may reduce deaths from breast cancer by half in women with ATM, CHEK2 and PALB2 gene mutations. The data suggests starting with annual breast MRIs between the ages of 30 to 35, and adding annual mammograms starting at age 40. Read the full article to learn …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post40122/
ICARE Social Media Post March 2022
BRCA1/2: Asian Breast Cancer Patients
A new study highlights the importance of customizing mutation carrier prediction models in order to improve the accuracy of predicting the likelihood of carrying a BRCA mutation in Asian breast cancer patients.Read the article for more info!https://ascopubs.org/doi/abs/10.1200/JCO.21.01647?cid=DM9826&bid=143994923Reference: Hong Ang et al. J Clin Oncol. 2022 Feb 10;JCO2101647. PMID: 35143328.
Permanent link to this article: https://inheritedcancer.net/post32522/
ICARE Social Media Post March 2022
Breast Cancer: Leading Cause Of Cancer-Associated Death Among Black Women
Did you know breast cancer surpassed lung cancer as the leading cause of cancer death among Black women in the United States? Although Black women are at a lower risk for developing breast cancer, they are 41% more likely to die of breast cancer compared with White women.Future research is needed to reverse course, through …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post31722/
ICARE Social Media Post March 2022
FDA Approves Olaparib for Adjuvant Treatment of High-risk Early Breast Cancer
On March 11th, the Food and Drug Administration (FDA) approved olaparib (Lynparza) for the adjuvant treatment of BRCA1/2 carriers with human epidermal growth factor receptor 2 (HER2)-negative, high-risk early breast cancer who have been treated with neoadjuvant or adjuvant chemotherapy.Learn more at 👇https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-adjuvant-treatment-high-risk-early-breast-cancer
Permanent link to this article: https://inheritedcancer.net/post31522/
ICARE Social Media Post March 2022
Komen Blog Post: Dr. Pal Interview
Komen Scholar, ICARE Founder and Clinical Geneticist, Dr. Tuya Pal, was recently interviewed for the Komen Blog. In the interview, Dr. Pal discusses the importance of making genetic counseling and testing more accessible in order to help populations who are at high risk for inherited cancer make more informed decisions about their medical care.Read the …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post30922/
ICARE Social Media Post March 2022
Lynch Syndrome: Uterine Cancer Treatment
A new study indicated treatment with pembrolizumab (Ketruda) resulted in benefit in patients with MSI-H advanced uterine cancer. Read the full article to learn more!https://ascopubs.org/doi/10.1200/JCO.21.01874Reference: O’Malley et al. J Clin Oncol. 2022 Jan 6;JCO2101874. PMID: 34990208.
Permanent link to this article: https://inheritedcancer.net/post30122/
ICARE Social Media Post February 2022
Use of Germline BRCA Testing in Patients With Ovarian Cancer and Commercial Insurance
A recent study found that only about 33% of women with ovarian cancer undergo germline BRCA1/2 testing, despite universal recommendations for such patients to have germline genetic testing. Check out the article for more information!https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2787937?resultClick=3Reference: Cham et al. JAMA Netw Open. 2022 Jan 4;5(1):e2142703. PMID: 35015069.
Permanent link to this article: https://inheritedcancer.net/post22122/
ICARE Social Media Post Month Year
Genetic Testing & Mortality Among Women with Breast or Ovarian Cancer
A recent study found there were BETTER short-term outcomes among women with:• triple-negative breast cancer and BRCA1 or BRCA2 mutations• ovarian cancer and BRCA2, BRIP1, RAD51C, or ATM mutationsThese findings may be reassuring to individuals with inherited gene mutations related to breast and ovarian cancer. Read the article to learn more!https://pubmed.ncbi.nlm.nih.gov/34373918/Reference: Kurian et al. J …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post21822/
ICARE Social Media Post February 2022
BRCA1/2 Cancer Risks: Oral Contraceptives
A new study found that among BRCA1/2 carriers oral contraceptive use strongly decreases lifetime cancer risk, despite an 𝗶𝗻𝗶𝘁𝗶𝗮𝗹 increase in breast, ovarian, and endometrial cancer risk. Read the full article to learn more!https://pubmed.ncbi.nlm.nih.gov/35048954/Reference: Schrijver et al. J Natl Cancer Inst. 2022 Jan 20;djac004. PMID: 35048954
Permanent link to this article: https://inheritedcancer.net/post21522/
Permanent link to this article: https://inheritedcancer.net/post20822/
ICARE Social Media Post February 2022
Breast Cancer Characteristics Across Genes
A new study demonstrated that breast cancer pathology and other clinical features differ by inherited breast cancer gene. Read the full article to learn more!https://jamanetwork.com/journals/jamaoncology/fullarticle/2788577Reference: Breast Cancer Association Consortium. JAMA Oncol. 2022 Jan 27. PMID: 35084436
Permanent link to this article: https://inheritedcancer.net/post20122/
ICARE Social Media Post January 2022
Male Cancer Risks: BRCA1 & BRCA2
A new study found that male BRCA1/2 carriers have a higher risk for breast, pancreatic, and stomach cancer. Additionally, male BRCA2 carriers were found to have higher risks for prostate cancer. Read the full Journal of Clinical Oncology article to learn more!https://ascopubs.org/doi/full/10.1200/JCO.21.02112Reference: Li et al. J Clin Oncol. 2022 Jan 25;JCO2102112. PMID: 35077220.
Permanent link to this article: https://inheritedcancer.net/post12822/
ICARE Social Media Post January 2022
Overview of genetic services delivery for hereditary breast cancer
A recent article by Drs. Sonya Reid, Tuya Pal, and Lucy Spalluto, and Katie Lang, MS, CGC and Anne Weidner, MPH of the ICARE team, has just been published with @SpringerNature in 𝘉𝘳𝘦𝘢𝘴𝘵 𝘊𝘢𝘯𝘤𝘦𝘳 𝘙𝘦𝘴𝘦𝘢𝘳𝘤𝘩 𝘢𝘯𝘥 𝘛𝘳𝘦𝘢𝘵𝘮𝘦𝘯𝘵. This article outlines the importance of genetic services and follow-up care for hereditary breast cancer, as well as …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12622/
ICARE Social Media Post January 2022
BRCA Breast Cancer Risks After Age 60
A study that included ICARE participants reported the risk of developing breast cancer remains high after age 60 in both BRCA1 and BRCA2 carriers. Read the article to learn more!https://pubmed.ncbi.nlm.nih.gov/33423179/Reference: Stjepanovic et al. Breast Cancer Res Treat. 2021 Jun;187(2):515-523. PMID: 33423179
Permanent link to this article: https://inheritedcancer.net/post12522/
ICARE Social Media Post January 2022
Belzutifan in Von Hippel-Lindau Disease Patients
Belzutifan demonstrated clinical efficacy in patients with renal cell carcinoma who have Von Hippel-Lindau Disease. Learn more by reading the full The New England Journal of Medicine article at 👇https://www.nejm.org/doi/full/10.1056/NEJMoa2103425Reference: Jonasch, et al. N Engl J Med. 2021 Nov 25;385(22):2036-2046. PMID: 34818478.
Permanent link to this article: https://inheritedcancer.net/post12122/
ICARE Social Media Post January 2022
Dr. Pal’s GIM Editorial
Estimating Polygenetic Risk Scores in non-Europeans through extrapolating from data in European descent populations is not accurate! Diversity in genomic studies is critically needed for both equity and scientific discovery. For more information, read the Genetics in Medicine editorial by ICARE Founder, Dr. Tuya Pal, here 👇https://www.gimjournal.org/article/S1098-3600(21)05396-X/fulltextReference: Pal. Genet Med. 2021 Nov 20;S1098-3600(21)05396-X. PMID: 34906472.
Permanent link to this article: https://inheritedcancer.net/post11822/
ICARE Social Media Post January 2022
Gastric Cancer Risks: CDH1
Read the 𝘑𝘈𝘔𝘈 𝘚𝘶𝘳𝘨𝘦𝘳𝘺 article to learn morehttps://jamanetwork.com/journals/jamasurgery/article-abstract/2785097Reference: Gamble et al. JAMA Surg. 2021 Oct 13;e215118. PMID: 34643667.
Permanent link to this article: https://inheritedcancer.net/post11122/
ICARE Social Media Post January 2022
Ovarian Cancer: BRCA1/2 Maintenance Olaparib
Read the full article for more details 👇https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(21)00531-3/fulltextReference: Banerjee et al. Lancet Oncol. 2021 Dec;22(12):1721-1731. PMID: 34715071.
Permanent link to this article: https://inheritedcancer.net/post10422/
ICARE Social Media Post December 2021
Fall 2021 Community Spotlight
In every ICARE newsletter we feature a 𝗰𝗼𝗺𝗺𝘂𝗻𝗶𝘁𝘆 𝘀𝗽𝗼𝘁𝗹𝗶𝗴𝗵𝘁 to share their experience with inherited cancer. In the latest edition, Dan Dry Dock Shockley, shares his experience with attenuated familial adenomatous polyposis (AFAP). Check out his full story at 👇https://inheritedcancer.net/community-spotlight/
Permanent link to this article: https://inheritedcancer.net/post123021/
ICARE Social Media Post December 2021
Fall 2021 Ask the Expert
In every ICARE newsletter we give our participants the opportunity to have a question addressed by an expert in the field. In the latest edition, Dr. Kerry Schaffer discusses the use of PARP inhibitors to treat inherited forms of prostate cancer.Check out Dr. Schaffer’s full response at 👇https://inheritedcancer.net/newsletters/
Permanent link to this article: https://inheritedcancer.net/post122821/
ICARE Social Media Post December 2021
Breast Cancer Risk Prediction Model for Black Women
A new breast cancer model has been developed and validated for breast cancer risk prediction, specifically for Black women in the United States. This is in contrast to prior models, which were developed in White women, and used in Black women (which UNDERPREDICTED risks). This is a notable advance as we strive towards health equity …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post122321/
ICARE Social Media Post December 2021
Li-Fraumeni Syndrome Cancer Risks
• Several variants were identified multiple times in those who did and did not meet Li-Fraumeni Syndrome clinical criteria: p.R175, p.G245, p.R248, p.R273, and p.R282• Other variants were exclusively found in those in the Li-Fraumeni Syndrome group: p.M133T, p.P152L, p.C275Y, p.C275, p.R337C, p.R342, and p.R342P• One variant was exclusively found in patients with attenuated Li-Fraumeni …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post122121/
ICARE Social Media Post December 2021
Prostate Cancer Screening: MSH2 & MSH6
A new study suggests men with MSH2 and MSH6 mutations have a higher incidence of prostate cancer, and may be candidates for PSA screening. Read the full article to learn more 👇 https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(21)00522-2/fulltext Reference: Bancroft et al. Lancet Oncol. 2021 Nov;22(11):1618-1631. PMID: 34678156.
Permanent link to this article: https://inheritedcancer.net/post121721/
ICARE Social Media Post December 2021
FDA Approves Belzutifan for Cancers Associated With von Hippel-Lindau Disease
On August 13th, the FDA approved the use of belzutifan (Welireg) for adult patients with von Hippel-Lindau (VHL) disease who require therapy for associated renal cell carcinoma, central nervous system hemangioblastomas, or pancreatic neuroendocrine tumors not requiring immediate surgery. For more information, check out The ASCO Post article at 👇https://ascopost.com/news/august-2021/fda-approves-belzutifan-for-cancers-associated-with-von-hippel-lindau-disease/?utm_source=TAP-EN-081321&utm_medium=email&utm_term=49cf1c97d48c2cf8231827e3bcb15769
Permanent link to this article: https://inheritedcancer.net/post121021/
ICARE Social Media Post December 2021
FDA Approves PARP Inhibitor (Olaparib) Treatment for some BRCA carriers with Early Stage Breast Cancer
FDA granted priority review to Olaparib for adjuvant treatment of certain patients with high-risk breast cancer. This designation applies to use of the agent by patients with BRCA-mutated, HER2-negative, high-risk early breast cancer who receive chemotherapy before or after surgery. For additional information, visit: https://tinyurl.com/healioFDAapproval
Permanent link to this article: https://inheritedcancer.net/post120621/
ICARE Social Media Post December 2021
The Impact of Subtype and Race on Breast Cancer Survival
A recent article 𝗹𝗲𝗱 𝗯𝘆 𝗗𝗿𝘀. 𝗦𝗼𝗻𝘆𝗮 𝗥𝗲𝗶𝗱 𝗮𝗻𝗱 𝗧𝘂𝘆𝗮 𝗣𝗮𝗹 𝗼𝗳 𝘁𝗵𝗲 𝗜𝗖𝗔𝗥𝗘 𝘁𝗲𝗮𝗺, published in 𝘉𝘳𝘦𝘢𝘴𝘵 𝘊𝘢𝘯𝘤𝘦𝘳 𝘙𝘦𝘴𝘦𝘢𝘳𝘤𝘩 𝘢𝘯𝘥 𝘛𝘳𝘦𝘢𝘵𝘮𝘦𝘯𝘵, outlines the impact of race and molecular subtype on HR+, HER2- breast cancer survival. Check out the full article at https://link.springer.com/content/pdf/10.1007/s10549-021-06342-0.pdf Reference: Reid et al. Breast Cancer Res Treat. 2021 Oct;189(3):845-852. PMID: 34331630.
Permanent link to this article: https://inheritedcancer.net/post120321/
ICARE Social Media Post November 2021
Variation in Colorectal Cancer Risk in Families With Lynch Syndrome
For more information, read the Lancet Oncology article led by The International Mismatch Repair Consortium at the below link 👇https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(21)00189-3/fulltext Reference: International Mismatch Repair Consortium. Lancet Oncol. 2021 Jul;22(7):1014-1022. PMID: 34111421.
Permanent link to this article: https://inheritedcancer.net/post113021/
ICARE Social Media Post November 2021
Changes in Racial and Ethnic Disparities in Access to Care and Health Among US Adults at Age 65 Years
For more information, view the article at 👇https://academic.oup.com/jnci/advance-article/doi/10.1093/jnci/djab141/6323231 Reference: Johnson et al. PMID: 34291289.
Permanent link to this article: https://inheritedcancer.net/post110921/
Permanent link to this article: https://inheritedcancer.net/post110221/
ICARE Social Media Post October 2021
Pancreatic Cancer Risks: ATM
For more information, read the full article at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2783767 Reference: Hsu, et al. JAMA Oncol. 2021 Sep 16:e213701. PMID: 34529012.
Permanent link to this article: https://inheritedcancer.net/post100121/
ICARE Social Media Post September 2021
USA Today Article: Fighting Cancer with Your Own Family History
Check out the full 𝘜𝘚𝘈 𝘛𝘰𝘥𝘢𝘺 article, featuring commentary from Dr. Tuya Pal (ICARE Founder), highlighting the importance of PALB2 as an inherited breast cancer gene: https://www.futureofpersonalhealth.com/breast-health/fighting-cancer-with-your-own-family-history/# Additional guidance is available through an impactful PALB2 practice resource recently published through ACMG: https://www.acmg.net/PDFLibrary/41436_2021_1151_OnlinePDF.pdf Reference: Tischkowitz, et al. Genet Med. 2021 Aug;23(8):1416-1423. PMID: 33976419
Permanent link to this article: https://inheritedcancer.net/post92221/
ICARE Social Media Post September 2021
New NCCN Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Guidelines (Released August 11th)
The National Comprehensive Cancer Network (NCCN) released new guidelines on August 11th for Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. You can check out the full guidelines by creating a FREE account at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf
Permanent link to this article: https://inheritedcancer.net/post91021/
ICARE Social Media Post August 2021
Genetic Risk Assessment for Hereditary Renal Cell Carcinoma
To view the 30 consensus statements, read the full article at: https://acsjournals.onlinelibrary.wiley.com/doi/10.1002/cncr.33679
Permanent link to this article: https://inheritedcancer.net/post83121/
ICARE Social Media Post August 2021
Breast Cancer Risks: BRCA1/2 & PALB2
For more information, view the article at 👇https://ascopubs.org/doi/full/10.1200/JCO.21.00531
Permanent link to this article: https://inheritedcancer.net/post82421/
ICARE Social Media Post August 2021
Metastatic Breast Cancer Genes
For additional information, read the Journal of Clinical Oncology article at the link below 👇 https://ascopubs.org/doi/10.1200/JCO.20.01200
Permanent link to this article: https://inheritedcancer.net/post82021/
ICARE Social Media Post August 2021
New York Times PALB2 Article
A New York Times article just published focused on the importance of PALB2 as a breast cancer gene (https://www.nytimes.com/…/breast-cancer-palb2-brca.html), which referenced our recent article focused on managing PALB2 carriers sponsored by ACMG – American College of Medical Genetics and Genomics, and developed through a worldwide collaboration (including: Marc Tischkowicz, Judith Balmana, Will Foulkes, Paul James, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post81921/
ICARE Social Media Post August 2021
Germline Testing & Advanced Cancers
Check out the article for more information: https://ascopubs.org/doi/pdf/10.1200/JCO.20.03661
Permanent link to this article: https://inheritedcancer.net/post81621/
ICARE Social Media Post August 2021
Breast Cancer Risk Among Breast Cancer Gene Carriers Varies by Polygenic Risk Score
For more information, read the 𝘑𝘰𝘶𝘳𝘯𝘢𝘭 𝘰𝘧 𝘊𝘭𝘪𝘯𝘪𝘤𝘢𝘭 𝘖𝘯𝘤𝘰𝘭𝘰𝘨𝘺 article at the link below:https://ascopubs.org/doi/figure/10.1200/JCO.20.01992
Permanent link to this article: https://inheritedcancer.net/post81321/
ICARE Social Media Post August 2021
ASCO Guideline Update: Olaparib for Breast Cancer
For additional information, read the updated American Society of Clinical Oncology (ASCO) recommendation (released June 15th, 2021) at the following link: https://www.asco.org/practice-patients/guidelines/breast-cancer?intcmp=ws_ascoorg_gdlns_hereditarybreastcancer_site_pressrelease_061621____#/143725
Permanent link to this article: https://inheritedcancer.net/post81021/
ICARE Social Media Post August 2021
Oral Contraceptive Use & Reduced Ovarian Cancer Risk in BRCA Carriers
For more information, view the AJOG article by Lieske Schrijver and colleagues at the following link: https://www.ajog.org/article/S0002-9378(21)00038-7/fulltext
Permanent link to this article: https://inheritedcancer.net/post80621/
ICARE Social Media Post August 2021
BRCA Mutations in Indian Women with Ovarian Cancer
For more information, view the article at: https://ascopubs.org/doi/full/10.1200/GO.21.00051
Permanent link to this article: https://inheritedcancer.net/post80321/
ICARE Social Media Post July 2021
Inherited Cancer in Colorectal Cancer Patients
For more information, view the article at: https://ascopubs.org/doi/10.1200/PO.20.00525
Permanent link to this article: https://inheritedcancer.net/post72721/
ICARE Social Media Post July 2021
Lifestyle & Colorectal Cancer Risk
For additional information, read the article at the link below: https://academic.oup.com/ajcn/article-abstract/113/4/810/6155851
Permanent link to this article: https://inheritedcancer.net/post72021/
ICARE Social Media Post July 2021
Niraparib Maintenance in BRCA Carriers with Ovarian Cancer
For more information visit: https://www.cancernetwork.com/view/multi-trial-analysis-indicates-niraparib-maintenance-for-brca-ovarian-cancer-leads-to-pfs-benefit-across-settings
Permanent link to this article: https://inheritedcancer.net/post71621/
ICARE Social Media Post July 2021
Disparities in Cancer Survival Among Adolescents and Young Adults
Check out the full article for more information: https://academic.oup.com/jnci/advance-article/doi/10.1093/jnci/djab006/6117446
Permanent link to this article: https://inheritedcancer.net/post71321/
ICARE Social Media Post July 2021
RAD51C & RAD51D Response to Rucaparib in Ovarian Cancer
Check out the Nature Communications article for more information: https://www.nature.com/articles/s41467-021-22582-6
Permanent link to this article: https://inheritedcancer.net/post70921/
ICARE Social Media Post July 2021
Advances in Pancreatic Cancer Treatment: PALB2 & BRCA1/2
For more information, view the article at the following link below: https://ascopubs.org/doi/abs/10.1200/JCO.21.00003 You may also read the ASCO post article at: https://ascopost.com/news/may-2021/maintenance-rucaparib-in-patients-with-platinum-sensitive-pancreatic-cancer-and-germline-or-somatic-brca1-brca2-or-palb2-variants/?utm_source=TAP%2DEN%2D051221%2DTrending%5FLymphoma&utm_medium=email&utm_term=49cf1c97d48c2cf8231827e3bcb15769
Permanent link to this article: https://inheritedcancer.net/post70621/
ICARE Social Media Post July 2021
PALB2 & Prostate Cancer
For more information, read the article available at: https://www.nature.com/articles/s41416-021-01410-0
Permanent link to this article: https://inheritedcancer.net/post70221/
ICARE Social Media Post June 2021
BRCA: Better Survival with Screening
For more information, please visit: https://www.cancertherapyadvisor.com/home/news/conference-coverage/st-gallen-international-breast-cancer-conference/sg-bcc-2021/breast-cancer-screening-brca-improves-survival-mutation
Permanent link to this article: https://inheritedcancer.net/post62921/
ICARE Social Media Post June 2021
A Population-Based Study of Genes Previously Implicated in Breast Cancer
For additional information, read the article at the following link: https://www.nejm.org/doi/10.1056/NEJMoa2005936
Permanent link to this article: https://inheritedcancer.net/post62521/
ICARE Social Media Post June 2021
BRCA2 Variants of Uncertain Significance
For more information, view the article at: https://pubmed.ncbi.nlm.nih.gov/33609447/
Permanent link to this article: https://inheritedcancer.net/post62221/
Permanent link to this article: https://inheritedcancer.net/post61521/
Permanent link to this article: https://inheritedcancer.net/post61121/
ICARE Social Media Post May 2021
Race-Based Oncology
For more information, view the article at the following link: https://ascopubs.org/doi/full/10.1200/PO.20.00418
Permanent link to this article: https://inheritedcancer.net/post52821/
ICARE Social Media Post May 2021
Breast Cancer Awareness Medical Minute
In recognition of 𝗪𝗼𝗺𝗲𝗻’𝘀 𝗛𝗲𝗮𝗹𝘁𝗵 𝗠𝗼𝗻𝘁𝗵, listen to our colleague, Dr. Sonya Reid, talk about the importance of mammography for the early detection of breast cancer! To learn more, check out the American Cancer Society’s Recommendations for the Early Detection of Breast Cancer at: https://www.cancer.org/cancer/breast-cancer/screening-tests-and-early-detection/american-cancer-society-recommendations-for-the-early-detection-of-breast-cancer.html
Permanent link to this article: https://inheritedcancer.net/post52421/
ICARE Social Media Post May 2021
USPSTF Earlier Colorectal Cancer Screening At Age 45
The United States Preventive Services Task Force (USPSTF) now recommends routine colorectal cancer screening begin at age 45 (lowered from the previous recommendation to start screening at age 50) due to an increasing number of colorectal cancer cases in younger adults. For more information, check out the full JAMA article at: https://jamanetwork.com/journals/jama/fullarticle/2779985
Permanent link to this article: https://inheritedcancer.net/post52121/
ICARE Social Media Post May 2021
ACMG Gene List
The American College of Medical Genetics and Genomics (ACMG) recently released the highly anticipated recommended minimum gene list for reporting secondary findings (SF) in clinical exome and genome sequencing. ACMG SF v3.0 added 3 new cancer genes (𝘗𝘈𝘓𝘉2, 𝘔𝘈𝘟, and 𝘛𝘔𝘌𝘔127) to bring the total number of inherited cancer genes to 28. For more information …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post52121_2/
ICARE Social Media Post May 2021
Plexiform Neurofibromas and NF1
For more information, read the article available at: https://ascopubs.org/doi/full/10.1200/JCO.20.02220
Permanent link to this article: https://inheritedcancer.net/post51821/
ICARE Social Media Post May 2021
CDKN2A: Cancer Risks and Risk Management
𝘐𝘯 𝘳𝘦𝘤𝘰𝘨𝘯𝘪𝘵𝘪𝘰𝘯 𝘰𝘧 𝘴𝘬𝘪𝘯 𝘤𝘢𝘯𝘤𝘦𝘳 𝘢𝘸𝘢𝘳𝘦𝘯𝘦𝘴𝘴 𝘮𝘰𝘯𝘵𝘩, we present cancer risks and management for 𝗖𝗗𝗞𝗡𝟮𝗔 per National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Version 2.2021 𝗠𝗲𝗻 & 𝗪𝗼𝗺𝗲𝗻:Melanoma risk: Elevated at 28-67% – Recommend annual full-body skin exam, regular self-examinations, and routine sun protective behaviors. Pancreatic cancer risk: >15% – Consider MRI/MRCP and/or endoscopic …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post51321/
ICARE Social Media Post May 2021
ACMG Clinical Practice Resource on PALB2
Check out the newly released ACMG Clinical Practice Resource on 𝗣𝗔𝗟𝗕𝟮 developed through a group of worldwide experts!https://www.acmg.net/PDFLibrary/41436_2021_1151_OnlinePDF.pdf Read the ACMG press release at: https://www.acmg.net/PDFLibrary/Global%20Team%20of%20Cancer%20Genetic%20Specialists%20release%20final%20template%205%206.final.pdf
Permanent link to this article: https://inheritedcancer.net/post51121/
ICARE Social Media Post May 2021
Direct-to-Consumer Tests
For additional information, view the BMJ article available at: 👉 https://www.bmj.com/content/372/bmj.n214
Permanent link to this article: https://inheritedcancer.net/post50321/
ICARE Social Media Post April 2021
Reducing Hereditary Cancer Act
The 𝙍𝙚𝙙𝙪𝙘𝙞𝙣𝙜 𝙃𝙚𝙧𝙚𝙙𝙞𝙩𝙖𝙧𝙮 𝘾𝙖𝙣𝙘𝙚𝙧 𝘼𝙘𝙩 is being proposed to ensure Medicare beneficiaries have access to inherited cancer genetic testing, increased screening, and risk-reducing procedures, when medically necessary & appropriate. Under current Medicare guidelines, only those with “signs, symptoms, complaints, or personal histories of disease” meet the criteria for medical services coverage. Genetic testing is only …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post43021/
ICARE Social Media Post April 2021
Effect of Salpingo-Oophorectomy on Breast Cancer Risk in Women With BRCA1 or BRCA2 Pathogenic Variants
For more information, view the JAMA Oncology article available at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2776761
Permanent link to this article: https://inheritedcancer.net/post42821/
ICARE Social Media Post April 2021
Addressing Racial Disparities in Breast Cancer Clinical Trial Enrollment
For more information, read the recently published ASCO Daily News editorial led by 𝗗𝗿. 𝗦𝗼𝗻𝘆𝗮 𝗥𝗲𝗶𝗱 from Vanderbilt University Medical Center, which focused on racial disparities in breast cancer clinical trial enrollment: https://dailynews.ascopubs.org/do/10.1200/ADN.21.200499/full/
Permanent link to this article: https://inheritedcancer.net/post42321/
ICARE Social Media Post April 2021
Komen Disparities Resource
Check out Susan G. Komen’s “𝗞𝗻𝗼𝘄 𝗬𝗼𝘂𝗿 𝗛𝗶𝘀𝘁𝗼𝗿𝘆, 𝗞𝗻𝗼𝘄 𝗬𝗼𝘂𝗿 𝗥𝗶𝘀𝗸” resource, which outlines disparities in breast cancer, how to understand your risk, and recommended screening and management practices. To learn more, please visit: 👉 https://www.komen.org/about-komen/our-impact/breast-cancer/health-equities-initiative/know-your-history/ 👉 https://blog.komen.org/news/black-history-month/
Permanent link to this article: https://inheritedcancer.net/post42021/
ICARE Social Media Post April 2021
Olaparib for Metastatic Breast Cancer
For more information, view the article at: https://ascopubs.org/doi/abs/10.1200/JCO.20.02151
Permanent link to this article: https://inheritedcancer.net/post41321/
ICARE Social Media Post April 2021
Winter 2021 Ask the Expert
In every ICARE newsletter we give our participants the opportunity to have a question addressed by an expert in the field. In the latest edition, Dr. Rebecca Smith explains how a genetic change (or mutation) is classified as pathogenic or benign. Check out Dr. Smith’s full response in our Winter 2021 newsletter at: https://inheritedcancer.net/newsletters/
Permanent link to this article: https://inheritedcancer.net/post40921/
ICARE Social Media Post April 2021
Fruits and Vegetables Lower Mortality
For further information, view the article available at:https://www.ahajournals.org/doi/abs/10.1161/CIRCULATIONAHA.120.048996
Permanent link to this article: https://inheritedcancer.net/post40621/
ICARE Social Media Post April 2021
Family Communication of Genetic Test Results Among Women with Inherited Breast Cancer Genes
Check out a recent article led by the ICARE team, published in the Journal of Genetic Counseling, outlining family communication of genetic test results among female BRCA1/2, PALB2, CHEK2, and ATM carriers. Check out the full article at: https://onlinelibrary.wiley.com/doi/full/10.1002/jgc4.1356
Permanent link to this article: https://inheritedcancer.net/post40221/
ICARE Social Media Post March 2021
STK11: Cancer Risks and Risk Management
Gene: STK11 Cancer Risks and Management per National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal Version 1.2020 Women:Breast cancer risk: Elevated at 40-60% – Recommend annual mammogram and breast MRI starting at around age 30. Ovarian tumor risk (typically benign sex cord/Sertoli cell tumors): Elevated at 18-21% – Recommend annual pelvic exam starting at …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post33021/
ICARE Social Media Post March 2021
MUTYH: Cancer Risks and Risk Management
Gene: MUTYH Cancer Risks and Management per National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal Version 1.2020 Men & women with two mutations in MUTYH:Colon cancer risk: High risk – Recommend colonoscopy every 1-2 years beginning at age 25-30; colectomy considered based on polyp burden and age. Duodenal cancer risk: Elevated – Consider baseline …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post32321/
ICARE Social Media Post March 2021
Winter 2021 Community Spotlight
In every ICARE newsletter we feature a 𝗰𝗼𝗺𝗺𝘂𝗻𝗶𝘁𝘆 𝘀𝗽𝗼𝘁𝗹𝗶𝗴𝗵𝘁 to share their experience with inherited cancer. In the latest edition, Dave Dubin, co-founder of AliveAndKickn, shares his experience with Lynch Syndrome. Check out his full story at: https://inheritedcancer.net/community-spotlight/
Permanent link to this article: https://inheritedcancer.net/post31921/
ICARE Social Media Post March 2021
APC: Cancer Risks and Risk Management
𝗚𝗲𝗻𝗲: 𝗔𝗣𝗖 Cancer Risks and Management per National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal Version 1.2020 𝗠𝗲𝗻 & 𝗪𝗼𝗺𝗲𝗻:𝘈𝘗𝘊 mutation leading to classic form of Familial Adenomatous Polyposis (FAP):Colorectal cancer risk: >99% if untreated – Treatment is based on polyp burden and includes proctocolectomy (with subsequent endoscopic screening of the ileal pouch) or …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post31621/
ICARE Social Media Post March 2021
Colorectal Cancer Risk Assessment Tool
In honor of 𝗖𝗼𝗹𝗼𝗿𝗲𝗰𝘁𝗮𝗹 𝗖𝗮𝗻𝗰𝗲𝗿 𝗔𝘄𝗮𝗿𝗲𝗻𝗲𝘀𝘀 𝗠𝗼𝗻𝘁𝗵, we wanted to highlight the National Cancer Institute’s Colorectal Cancer Risk Assessment Tool (CCRAT) designed for healthcare providers to use with select patients to estimate the risk of colorectal cancer. You may access the tool at https://ccrisktool.cancer.gov
Permanent link to this article: https://inheritedcancer.net/post31221/
ICARE Social Media Post March 2021
Three Articles: Breast Cancer Risks
For additional information about the: 》US-based study, visit: https://www.nejm.org/doi/full/10.1056/nejmoa2005936 》International study, visit: https://www.nejm.org/doi/full/10.1056/nejmoa1913948 》Accompanying editorial: https://www.nejm.org/doi/full/10.1056/NEJMe2035083
Permanent link to this article: https://inheritedcancer.net/post30921/
ICARE Social Media Post March 2021
Breastfeeding and the Risk of Epithelial Ovarian Cancer Among Women with A BRCA1 or BRCA2 Mutation
For further information, view the article available at: https://www.gynecologiconcology-online.net/action/showPdf?pii=S0090-8258%2820%2933947-0
Permanent link to this article: https://inheritedcancer.net/post30221/
ICARE Social Media Post February 2021
Olaparib and Invasive Disease-Free Survival in Patients with BRCA, High-Risk, Early Breast Cancer
For additional information visit: https://www.onclive.com/view/adjuvant-olaparib-showcases-idfs-improvement-in-brca-high-risk-early-breast-cancer
Permanent link to this article: https://inheritedcancer.net/post22321/
ICARE Newsletter Winter 2021
Inherited Breast Cancer Genes: Two New Important Articles Just Released
Results of a United States (U.S.)-based study1 and an international study2 were released in January in the New England Journal of Medicine and provide a much clearer picture about the role of inherited breast cancer genes in women without a family history of cancer, and how common these genes may be in the general population. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nlw2021/
ICARE Newsletter Winter 2021
Updates to National Comprehensive Cancer Network (NCCN) Guidelines Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic
Released September 8th, 2020: Genetic testing criteria by cancer type: Breast Cancer: Broadened to include relatives with ALL grades of prostate cancer (not just high-grade) Having multiple breast cancer diagnoses no longer depends on whether the diagnoses were on two different breasts Prostate Cancer: Now includes cribriform histology and ANY risk group (not just high-grade …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nlw2021/
ICARE Newsletter Winter 2021
Exploring Disparities Among Those with Inherited Cancers
It has never been more urgent to ensure that advances in genomic technologies do not further widen existing cancer health disparities. In the fall of 2020, the American Association for Cancer Research (AACR) put forth a report focused on cancer health disparities, in which they highlighted several issues.1 Notably, disparities in inherited cancer care were …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nlw2021/
ICARE Newsletter Winter 2021
Study Suggests Low Yield of MRI Surveillance After Bilateral Mastectomy
A study of 159 women, including BRCA1/2 carriers, who had a bilateral mastectomy with reconstruction and underwent breast MRI screening, showed few women had detection of breast cancer through MRI after their bilateral mastectomy. These results support the recommendation that BRCA1/2 carriers with or without breast cancer who have a bilateral mastectomy with reconstruction do …
Continue reading
Permanent link to this article: https://inheritedcancer.net/8nlw2021/
ICARE Newsletter Winter 2021
Community Spotlight

My genes don’t define me. I am AliveAndKickn. Pretty bold statement. AliveAndKickn is more than just a name. It’s a way of life. I joke that Lynch Syndrome is the genetic predisposition to colon cancer, endometrial cancer, other cancers…and soccer. But that’s just me. Besides half a dozen surgeries since 1997, I have and still play and coach the game I love. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/spotlightnlw2021/
ICARE Newsletter Winter 2021
Learning You Have a Mutation in an Inherited Cancer Gene: What’s Next?
The benefits achieved through genetic testing for inherited cancer only happen by acting upon the results. This can be through guiding cancer treatment, receiving appropriate cancer risk management strategies, and sharing results with at-risk family members so they too can benefit from this information. We recently reported on results of our study, made possible through …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nlw2021/
ICARE Newsletter Winter 2021
CHEK2 is NOT a Li-Fraumeni Syndrome Gene
An old study back in 1999 suggested that CHEK2 may be a Li-Fraumeni Syndrome gene.1 However, a subsequent report in 2002 clearly refuted this original assertion, and based on additional data and analysis concluded that “… it is very unlikely that CHEK2 is an alternative Li-Fraumeni Syndrome susceptibility gene.”2 Another subsequent report in 2008 based …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nlw2021/
ICARE Newsletter Winter 2021
Assessing How Pregnancy and Breastfeeding May Affect Cancer Risks in BRCA Carriers
Results of a recently published study suggested that pregnancy after breast cancer in BRCA carriers does not lead to a worse outcome in women or their fetuses.1 This information is reassuring for BRCA carriers who have had a prior diagnosis of breast cancer and are considering having children. In another study among female BRCA carriers …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nlw2021/
ICARE Newsletter Winter 2021
Ask the Expert

In each newsletter, we give participants the opportunity to have their questions addressed by experts in the field. This question was addressed by Rebecca Smith, PhD, Laboratory Director at Genetics Associates, Inc. with over 20 years of experience in biomedical research and 7 years of experience in clinical laboratory diagnostics. If you have a question …
Continue reading
Permanent link to this article: https://inheritedcancer.net/9nlw2021/
ICARE Newsletter Winter 2021
Inherited Cancer Treatment: Updates and Relevant Policies
Over the last several months, the American Society of Clinical Oncology published a number of guidelines related to the use of PARP inhibitors among those with BRCA-associated cancers, including guidelines focused on ovarian cancer,1 metastatic pancreatic cancer,2 and breast cancer.3 Additionally, costs of drugs also have great potential to influence policy, highlighting the importance of …
Continue reading
Permanent link to this article: https://inheritedcancer.net/7nlw2021/
ICARE Social Media Post February 2021
ICARE Winter 2021 Newsletter
The ICARE Winter 2021 Newsletter is now available! Check out this latest edition for recent research and clinical updates, a Q&A with a genomics expert, and an inspiring community spotlight piece. You can read the newsletter by visiting: https://inheritedcancer.net/wp-content/uploads/ICARE-2021-Winter-Newsletter.pdf. Please feel free to share with family members, friends, and/or your healthcare providers.
Permanent link to this article: https://inheritedcancer.net/post20921/
ICARE Social Media Post February 2021
Sharing Genetic Test Results with Family Members of BRCA, PALB2, CHEK2, and ATM Carriers
Our team recently published “Sharing Genetic Test Results with Family Members of 𝘉𝘙𝘊𝘈, 𝘗𝘈𝘓𝘉2, 𝘊𝘏𝘌𝘒2, and 𝘈𝘛𝘔 Carriers” in 𝘗𝘢𝘵𝘪𝘦𝘯𝘵 𝘌𝘥𝘶𝘤𝘢𝘵𝘪𝘰𝘯 𝘢𝘯𝘥 𝘊𝘰𝘶𝘯𝘴𝘦𝘭𝘪𝘯𝘨 Special Issue on Genetics. View the article available at:https://www.sciencedirect.com/science/article/pii/S0738399120306832 Challenges and barriers to family sharing included concern for family members’ reactions, complexities of information, lack of closeness, perceived relevance, & emotional impact. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post20821/
ICARE Social Media Post February 2021
Strategies to Enhance the Identification of Hereditary Breast Cancer Gene Carriers
Check out a recent review article led by the ICARE team, published in 𝘌𝘹𝘱𝘦𝘳𝘵 𝘙𝘦𝘷𝘪𝘦𝘸 𝘰𝘧 𝘔𝘰𝘭𝘦𝘤𝘶𝘭𝘢𝘳 𝘋𝘪𝘢𝘨𝘯𝘰𝘴𝘵𝘪𝘤𝘴, outlining strategies to improve the identification of inherited breast cancer gene carriers. Check out the full article at: https://www.tandfonline.com/doi/pdf/10.1080/14737159.2020.1816829?needAccess=true
Permanent link to this article: https://inheritedcancer.net/post20221/
ICARE Social Media Post January 2021
Male Breast Cancer Global Alliance
The Male Breast Cancer Global Alliance shares stories from male breast cancer survivors all over the world. To learn more and find resources for affected men and their families, please visit www.mbcglobalalliance.org. The Male Breast Cancer Global Alliance also developed printable and shareable cards in over 20 languages outlining the proper way to do self-breast …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12621/
ICARE Social Media Post January 2021
COVID-19 Vaccine Myths
Although we don’t typically post on vaccines, given the public health importance of the COVID-19 vaccine and the questions we have received related to whether the vaccine could change one’s DNA, we …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12221/
ICARE Social Media Post January 2021
Sharing Genetic Test Results with Family Members of BRCA, PALB2, CHEK2, and ATM
Check out a new article by the ICARE team, published in Patient Education and Counseling, evaluating the motivators and barriers to sharing personal genetic test results with family members. The article is 𝗳𝗿𝗲𝗲 to access and download 𝘂𝗻𝘁𝗶𝗹 𝗠𝗮𝗿𝗰𝗵 𝟱𝘁𝗵 at: https://www.sciencedirect.com/science/article/pii/S0738399120306832
Permanent link to this article: https://inheritedcancer.net/post11521/
ICARE Social Media Post January 2021
Qualitative Methods for Refining a Web-Based Educational Tool for Patients Focused on Inherited Cancer Predisposition
Check out a study led by the ICARE team, published with @SpringerNature in the Journal of Cancer Education, outlining the evaluation and refinement strategies of an interactive web-based tool developed to provide education about inherited cancer risk. Check out a full-text view-only version of the paper at https://rdcu.be/cc8sR or read the abstract at https://pubmed.ncbi.nlm.nih.gov/33400205/ (PMID: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11221/
ICARE Social Media Post January 2021
Family Sharing Resources: GeneSHARE
With the tremendous advances in gene-based care among those at risk for inherited cancer, we are trying to develop and improve tools and strategies to make it easier for more people to benefit from genetic testing. We are excited to share with you a free online toolkit called 𝗚𝗲𝗻𝗲𝗦𝗛𝗔𝗥𝗘, which is aimed at helping patients …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post10821/
ICARE Social Media Post January 2021
National Cancer Institute (NCI) Cancer Genetics PDQ
For further information, check out the National Cancer Institute’s PDQ® Cancer Genetics Summary available at: https://www.cancer.gov/publications/pdq/information-summaries/genetics
Permanent link to this article: https://inheritedcancer.net/post10521/
ICARE Social Media Post December 2020
Genetic Testing in Women with Breast Cancer
Approximately 4,000 women with breast cancer were tested for mutations in nine breast cancer genes – 6.2% had mutations in at least one of the nine genes, and 2.7% had mutations in either 𝘽𝙍𝘾𝘼1 or 𝘽𝙍𝘾𝘼2. Comparisons between women who did versus did not meet National Comprehensive Cancer Network (NCCN) guidelines for testing showed that: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post122920/
Permanent link to this article: https://inheritedcancer.net/post122220/
ICARE Social Media Post December 2020
CHEK2 is NOT a Li-Fraumeni Syndrome Gene
For additional details, read the articles at the links below: 𝟭𝟵𝟵𝟵: https://pubmed.ncbi.nlm.nih.gov/10617473/ 𝟮𝟬𝟬𝟮: https://pubmed.ncbi.nlm.nih.gov/12442270/ 𝟮𝟬𝟬𝟴: https://pubmed.ncbi.nlm.nih.gov/18178638/
Permanent link to this article: https://inheritedcancer.net/post121820/
ICARE Social Media Post December 2020
CDH1 Cancer Risks
When a 𝘾𝘿𝙃1 mutation is identified, several factors should be considered to figure out what the cancer risks might be and what medical management should be advised. Specifically, differences in cancer risks among those with a 𝘾𝘿𝙃1 mutation are not well understood. Identifying a 𝘾𝘿𝙃1 mutation in families with hereditary diffuse gastric cancer (HDGC) …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post121520/
ICARE Social Media Post December 2020
PALB2-associated Metastatic Breast Cancer
For further information, view the article at: https://ascopubs.org/doi/abs/10.1200/JCO.20.02151
Permanent link to this article: https://inheritedcancer.net/post121120/
ICARE Social Media Post December 2020
New NCCN Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Guidelines
The National Comprehensive Cancer Network (NCCN) released new guidelines on November 20th for Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. You can check out the full guidelines by creating a FREE account at: https://www.nccn.org/…/physician_gls/pdf/genetics_bop.pdf
Permanent link to this article: https://inheritedcancer.net/post120820/
ICARE Social Media Post December 2020
Broader Germline Testing for Urothelial Cancer
The most common inherited form of urothelial cancers is Lynch Syndrome. However, a study showed that of 586 individuals with urothelial cancer, 80 had a mutation in an inherited cancer gene (14%). Mutations in several genes were observed; however, 𝙈𝙎𝙃2 and 𝘽𝙍𝘾𝘼2 were both significantly associated with urothelial cancer (odds ratio of 3.7). Confirmatory …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post120420/
Permanent link to this article: https://inheritedcancer.net/post120120/
Permanent link to this article: https://inheritedcancer.net/post112720/
Permanent link to this article: https://inheritedcancer.net/post112420/
ICARE Social Media Post November 2020
ASCO Guideline Updates: Breast Cancer
The American Society of Clinical Oncology (ASCO) published updated guidelines for the management of hereditary breast cancer for the following gene carriers: 𝘽𝙍𝘾𝘼1/2 • Consider breast-conserving therapy • Consider nipple-sparing mastectomy, if medically appropriate • Advanced breast cancer: ⫸ PARP inhibitors (olaparib, talazoparib) preferred over non-platinum single agent chemotherapy ⫸ Platinum agents are recommended …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post112020/
ICARE Social Media Post November 2020
ASCO Guidelines for Patients with Metastatic Pancreatic Cancer
For additional details, refer to the article available at: https://ascopubs.org/doi/full/10.1200/JCO.20.01364
Permanent link to this article: https://inheritedcancer.net/post111720/
ICARE Social Media Post November 2020
Polygenic Risk Scores in Predicting Breast Cancer Risk
For further information, view the article available at: https://ascopubs.org/doi/full/10.1200/PO.19.00360
Permanent link to this article: https://inheritedcancer.net/post111320/
ICARE Social Media Post November 2020
B-GREAT 2020 Newsletter
The B-GREAT 2020 Newsletter is now available! Check out this latest edition for research updates and information about racial inequalities in healthcare. You can read the newsletter by visiting: https://bgreatinitiative.inheritedcancer.net/wp-content/uploads/BGREAT-Newsletter-2020.pdf. Please feel free to share with family members, friends, and/or your healthcare providers. We will be publishing these newsletters twice a year starting in 2021. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post111020/
ICARE Social Media Post November 2020
Clinical Trial Participation Powers Patient’s Positive Attitude
Brooke Thomas has leaned on 12 years of experience as a medical social worker and found ways to stay positive and upbeat through it all – and she has a lot to be positive about these days, thanks to an amazing response to her treatment as part of a clinical trial at Vanderbilt-Ingram Cancer Center. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post110620/
ICARE Social Media Post November 2020
Identifying Individuals At-Risk for Inherited Cancer: Disparities Among Ovarian Cancer Patients
This study highlights disparities among ovarian cancer patients. For further information, view the article at: https://pubmed.ncbi.nlm.nih.gov/30964716/
Permanent link to this article: https://inheritedcancer.net/post110320/
ICARE Social Media Post October 2020
CDH1: Cancer Risks and Risk Management
Gene: 𝘾𝘿𝙃𝟭 Cancer Risks and Management per NCCN Genetic/Familial High-Risk Assessment: Breast/Ovarian/Pancreatic Version 1.2021 and Gastric Version 3.2020: 𝗪𝗼𝗺𝗲𝗻: Breast cancer risk: Elevated at 55% – Recommend annual mammogram starting at age 30; consider breast MRI with contrast starting at age 30. 𝗠𝗲𝗻 𝗮𝗻𝗱 𝗪𝗼𝗺𝗲𝗻: Stomach cancer risk: Elevated at 83% for women, and 67% …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post103020/
ICARE Social Media Post October 2020
Polygenic Risk Scores in Refining Breast Cancer Risks
For further information, view the article available at: https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2767768
Permanent link to this article: https://inheritedcancer.net/post102720/
ICARE Social Media Post October 2020
PTEN: Cancer Risks and Risk Management
Gene: 𝙋𝙏𝙀𝙉 Cancer Risks and Management per National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Breast/Ovarian/Pancreatic Version 1.2021: 𝗪𝗼𝗺𝗲𝗻: Breast cancer risk: Elevated at 85% – Recommend annual mammogram starting at age 30-35 (or 5-10 years before the earliest known breast cancer in the family); consider breast MRI with contrast starting at age 30-35; consider …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102320/
ICARE Social Media Post October 2020
Three Genetic Variants Identified That May Raise Breast Cancer Risks in Men
For more information, view the article available at: https://academic.oup.com/jnci/advance-article/doi/10.1093/jnci/djaa101/5885091
Permanent link to this article: https://inheritedcancer.net/post102020/
ICARE Social Media Post October 2020
TP53: Cancer Risks and Risk Management
Gene: 𝙏𝙋𝟱𝟯 Syndrome: Li-Fraumeni Cancer Risks and Management per National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Breast/Ovarian/Pancreatic Version 1.2021: 𝗪𝗼𝗺𝗲𝗻: Breast cancer risk: Elevated at 54% – Recommend clinical breast exam every 6-12 months starting at age 20, annual breast MRI with contrast starting at age 20, and annual mammogram starting at age 30; …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post101620/
ICARE Social Media Post October 2020
New ASCO Guidelines On Use Of PARP Inhibitors To Manage Ovarian Cancer
New guidelines for the use of PARP inhibitors to treat ovarian cancer among those with BRCA1 or BRCA2 mutations were published through the American Society of Clinical Oncology (ASCO) to guide providers about the role of this class of drugs in the management of this type of cancer. Link to the guidelines are available at: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post101320/
ICARE Social Media Post October 2020
CHEK2: Cancer Risks and Risk Management
Gene: 𝘾𝙃𝙀𝙆2 Cancer Risks and Management per National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal Version 1.2020 & Breast/Ovarian/Pancreatic Version 1.2021: 𝗪𝗼𝗺𝗲𝗻: Breast Cancer Risk: Elevated at 28-44% – Recommend annual mammogram starting at age 40 and consider annual breast MRIs with contrast starting at age 40. 𝗠𝗲𝗻 𝗮𝗻𝗱 𝗪𝗼𝗺𝗲𝗻: Colorectal Cancer Risk: Elevated …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post100920/
ICARE Social Media Post October 2020
Addition of Veliparib to Carboplatin/Paclitaxel in Previously Treated Patients With BRCA-Mutated Advanced Breast Cancer
Among BRCA carriers with metastatic breast cancer, the combination of veliparib AND chemotherapy with platinum-based agents (carboplatin) and taxanes (paclitaxel) led to a longer duration of progression free survival (disease that did not progress), compared to those treated with ONLY chemotherapy. To read the full article visit: https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(20)30447-2/fulltext [This finding was previously outlined last year …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post100620/
ICARE Social Media Post October 2020
ATM: Cancer Risks and Risk Management
Gene: 𝘼𝙏𝙈 Cancer Risks and Management (per NCCN Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Version 1.2021) 𝗪𝗼𝗺𝗲𝗻: Breast cancer risk: Elevated at 30% – Recommend annual mammogram starting at age 40 and consider annual breast MRIs starting at age 40. Ovarian cancer risk: Possibly increased, not well established – Manage based on family history. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post100220/
ICARE Social Media Post September 2020
Living with Lynch 2020 Virtual Patient Workshop
SJoin the Colon Cancer Coalition and AliveAndKickn for the Living with Lynch 2020 Virtual Patient Workshop on𝗙𝗿𝗶𝗱𝗮𝘆, 𝗢𝗰𝘁𝗼𝗯𝗲𝗿 𝟵𝘁𝗵 𝗳𝗿𝗼𝗺 𝟭𝗽𝗺-𝟱𝗽𝗺 𝗘𝗧 to hear unique patient perspectives and the latest information from experts on Lynch syndrome. Visit https://www.livingwithlynch.org/2020-living-with-lynch to register for this free workshop today!
Permanent link to this article: https://inheritedcancer.net/post92920/
ICARE Social Media Post September 2020
AACR Cancer Disparities Progress Report 2020
The American Association for Cancer Research (AACR) published a Cancer Disparities Progress Report. “… our limited knowledge of cancer biology in racial and ethnic minorities, including their inherited cancer predisposition and the genomic underpinnings of cancer initiation and progression, diminishes the potential of precision medicine in these populations.” This report discusses relevant information about inherited …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post92520/
ICARE Social Media Post September 2020
FDA Grants Breakthrough Therapy Status to MK-6482 for Treatment of Patients With VHL Disease-Associated Renal Cell Carcinoma
For additional information, please visit: https://www.onclive.com/view/fda-grants-breakthrough-status-to-mk-6482-for-select-vhl-disease-associated-rcc Also check out the abstract presented at the 2020 ASCO meeting: https://meetinglibrary.asco.org/record/185945/abstract
Permanent link to this article: https://inheritedcancer.net/post92220/
Permanent link to this article: https://inheritedcancer.net/post91820/
ICARE Social Media Post September 2020
ICARE Summer 2020 Newsletter
The ICARE Summer 2020 Newsletter is now available! Check out this latest edition for recent research and clinical updates as well as a Q&A with a nationally renowned clinical geneticist from Vanderbilt-Ingram Cancer Center. You can read the newsletter by visiting: https://inheritedcancer.net/wp-content/uploads/ICARE-2020-Summer-Newsletter.pdf Please feel free to share with family members, friends, and/or your …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post91620/
ICARE Social Media Post September 2020
NCCN Genetic Testing Criteria Updates by Cancer
The National Comprehensive Cancer Network (NCCN) released new guidelines on September 8th, 2020, which included updates to genetic testing criteria by cancer type as follows: Breast Cancer: Broadened to include relatives with ALL grades of prostate cancer (not just high-grade) Now having multiple breast cancer diagnoses is NOT dependent on whether the diagnoses were …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post91520/
ICARE Social Media Post September 2020
NCCN Breast Cancer Pathology Updates
The National Comprehensive Cancer Network (NCCN) released new guidelines on September 8th, 2020. Check out the full guidelines by creating a FREE account at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf
Permanent link to this article: https://inheritedcancer.net/post91420/
ICARE Social Media Post September 2020
NCCN Genetic Testing Choice Updates
The National Comprehensive Cancer Network (NCCN) released new guidelines on September 8th, 2020, which included updates to genetic testing choices and considerations as follows: Choice of multi-gene panel may be guided by genes included, analyses offered, and financial assistance programs available for family testing Significant limitations in interpretations of polygenic risk scores: NOT recommended for …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post91120/
ICARE Social Media Post September 2020
NCCN Breast Cancer Risk Management Updates by Gene
The National Comprehensive Cancer Network (NCCN) released new guidelines on September 8th, 2020, which included updates to breast cancer risk management recommendations by gene as follows: NBN – high-risk breast screening was removed as there is insufficient evidence to support high cancer risks BARD1 – added consideration for high-risk breast screening starting at age 40 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post91020/
ICARE Social Media Post September 2020
ACMG Guidelines for Genetic Testing for All Breast Cancer
Check out Vanderbilt’s article about genetic testing considerations at: https://discover.vumc.org/2020/02/genetic-testing-considerations-for-breast-cancer-patients/ Check out the original document from the American College of Medical Genetics and Genomics at: https://www.nature.com/articles/s41436-019-0712-x
Permanent link to this article: https://inheritedcancer.net/post90820/
ICARE Social Media Post September 2020
GREM1 Duplication
The GREM1 gene leads to hereditary mixed polyposis syndrome, characterized by multiple polyps of mixed pathology and high risks for colorectal cancer. A specific duplication in the 5′ regulatory region of the GREM1 gene has been found in a subset of Ashkenazi Jewish individuals with hereditary mixed polyposis syndrome; therefore, GREM1 genetic testing is …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post90420/
ICARE Social Media Post September 2020
Biallelic Mutations in BRCA1 Cause Fanconi Anemia
BRCA1 is not typically thought to be one of the genes which cause Fanconi anemia. However, rare cases of individuals with biallelic BRCA1 mutations (mutations in both copies of the gene) have been reported. One case involved a woman who had breast cancer at age 23, and was born with multiple birth defects consistent with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post90120/
ICARE Social Media Post August 2020
Older Women with Breast Cancer Have Higher Risks of Ovarian and Other Cancers
Among 4,500 post-menopausal women with breast cancer, 3.55% had a mutation in a gene associated with inherited breast cancer (3-fold higher than what was seen among women who were cancer-free). BRCA1/2 mutations were seen more frequently in women diagnosed with breast cancer at or below age 65 (2.21%) compared to those diagnosed after age 65 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post82820/
ICARE Social Media Post August 2020
Pheochromocytoma and Paraganglioma Genetic Testing
More than 40% of patients with pheochromocytoma or paraganglioma, regardless of age and family history, carry an inherited gene mutation. Given that gene mutation carriers may benefit from gene-specific cancer screening, all individuals diagnosed with one of these tumors should seek genetic counseling to discuss genetic testing options. Genes that predispose to pheochromocytoma and paraganglioma …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post82520/
ICARE Social Media Post August 2020
Optimal Timing of Prophylactic Gastrectomy for CDH1 Carriers
Individuals with a CDH1 mutation and classic personal and family history of diffuse gastric (stomach) cancer have a very high risk of developing and dying from stomach cancer, if they do not have preventive surgery to remove their stomach. A simulation study to assess prophylactic total gastrectomy (PTG; the removal of the stomach to prevent …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post82120/
ICARE Social Media Post August 2020
MRI Surveillance Has Very Low Yield After Bilateral Mastectomy and Reconstruction
A study of 159 women, including BRCA carriers, who had bilateral mastectomy with reconstruction, underwent breast MRI screening. The results showed few women had detection of breast cancer through MRI, after their bilateral mastectomy. These results support the recommendation against screening MRI in women who have had bilateral mastectomy with reconstruction due to a diagnosis …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post81820/
ICARE Newsletter Summer 2020
Treatment Advances Among Those with Neurofibromatosis Type 1

There continue to be ongoing advances in treatment studies among those with inherited cancer gene mutations, which are rapidly being followed by FDA approval for specific cancer treatments. Select studies and advances are summarized below: Neurofibromatosis Type 1 (NF1): The FDA granted selumetinib (a MEK inhibitor) breakthrough therapy designation for treatment of inoperable plexiform neurofibromas.
Permanent link to this article: https://inheritedcancer.net/8nls2020/
ICARE Newsletter Summer 2020
Treatment Advances Among Those with Lynch Syndrome

There continue to be ongoing advances in treatment studies among those with inherited cancer gene mutations, which are rapidly being followed by FDA approval for specific cancer treatments. Select studies and advances are summarized below: Lynch Syndrome: Colorectal Cancer: Among patients with MSI-H or MMR-deficient colorectal cancers (frequently seen among those with Lynch syndrome), pembrolizumab …
Continue reading
Permanent link to this article: https://inheritedcancer.net/7nls2020/
ICARE Newsletter Summer 2020
Ask the Expert

In each newsletter, we give participants the opportunity to have their questions addressed by experts in the field. This question was addressed by Georgia Wiesner, MD, MS, a nationally renowned clinical cancer geneticist, who is an Ingram Professor of Cancer Research, Professor of Medicine in the Division of Genetic Medicine, and the Director of the …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nls2020/
ICARE Newsletter Summer 2020
Identifying Individuals At-Risk for Inherited Cancer
We have known for a while that many people who have mutations in BRCA1/2 and other inherited cancer risk genes are unaware of their mutation as they have not yet had genetic testing. A recent study among women aged 20 or older living in California and Georgia, which included almost 80,000 breast cancer patients and …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nls2020/
ICARE Newsletter Summer 2020
Community Spotlight

PTEN is one of the body’s tumor suppressor genes, which controls cell growth. When a PTEN mutation is present, cells may grow uncontrollably, causing tumors to develop that may become cancerous. A patient born with a PTEN mutation is at high risk for developing breast, thyroid, kidney, colon, and endometrial cancer. My PTEN journey began …
Continue reading
Permanent link to this article: https://inheritedcancer.net/spotlightnls2020/
ICARE Newsletter Summer 2020
Treatment Advances Among Those with Von-Hippel Lindau (VHL) Disease
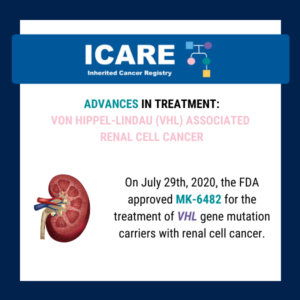
There continue to be ongoing advances in treatment studies among those with inherited cancer gene mutations, which are rapidly being followed by FDA approval for specific cancer treatments. Select studies and advances are summarized below: Von-Hippel Lindau (VHL) Disease: Among patients with VHL-associated clear cell renal cell carcinoma (RCC), a recent study suggested potential benefit …
Continue reading
Permanent link to this article: https://inheritedcancer.net/9nls2020/
ICARE Newsletter Summer 2020
Treatment Advances Among BRCA1/2 Carriers
There continue to be ongoing advances in treatment studies among those with inherited cancer gene mutations, which are rapidly being followed by FDA approval for specific cancer treatments. Select studies and advances are summarized below: BRCA1/2 Carriers: Breast Cancer: For those with later stage or metastatic breast cancer, the FDA currently has approvals for the use …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nls2020/
ICARE Newsletter Summer 2020
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal Guidelines

Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal Guidelines (Version 1.2020, posted July 21, 2020) For individuals with Lynch Syndrome: Cancer risks were updated based on information from recent studies: Main updates included cancer risks in PMS2 (endometrial, ovarian, and prostate cancer), MSH2 and EPCAM (prostate and brain cancer), and MSH6 (prostate …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nls2020/
ICARE Newsletter Summer 2020
Guideline-Concordant Care Among Women with Inherited Cancer Gene Mutations

Testing for inherited cancer among breast cancer patients has tremendous potential to guide appropriate care following testing. Yet, a number of efforts suggest that women are not consistently receiving care according to current national guidelines based on their genetic test result. In fact, results from studies suggest many women for whom risk-reducing mastectomy would not …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nls2020/
ICARE Newsletter Summer 2020
Polygenic Risk Scores and Colon Cancer

A recent study focused on how polygenic risk scores (PRS) may be related to colorectal cancers. Of note, PRS are calculated using genetic differences throughout someone’s DNA, in combination with their clinical and family history of cancer. PRS, alongside environmental and lifestyle risk factors, may help to identify people who may benefit from screening at …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nls2020/
ICARE Social Media Post August 2020
Inherited Prostate Cancer Risk
Over 600,000 men age 40 and older who were part of a family with at least three consecutive generations affected with prostate cancer were studied from the Utah Population Database. Findings from this study showed that: 36,000 had prostate cancer (5.9%) 2,500 had early-onset disease (7%) 4,000 had lethal disease (11.1%) 15,000 had clinically significant …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post81420/
ICARE Social Media Post August 2020
CDC Resource for Free Breast Cancer Screening
We are here to help! Our followers asked us to provide information about screening resources for women with hereditary breast cancer. We would like to make folks aware about a CDC program that provides free breast cancer screening for women who are uninsured and underinsured. Check out more information at: https://www.cdc.gov/cancer/nbccedp/
Permanent link to this article: https://inheritedcancer.net/post81120/
ICARE Social Media Post August 2020
Presymptomatic BRCA1/2 Carriers May Have Better Outcomes
Check out the article at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2768011
Permanent link to this article: https://inheritedcancer.net/post80720/
ICARE Social Media Post August 2020
Disparities in BRCA Counseling Across Diverse Populations
A recently published study based on data from our participants showed that few young Black women with breast cancer see a genetic counselor AND those with insurance are LESS likely to see a genetic counselor. For more information, check out the full study at: https://www.nature.com/articles/s41436-020-0762-0
Permanent link to this article: https://inheritedcancer.net/post80420/
ICARE Social Media Post August 2020
Genes That Predispose African American Women to Breast Cancer
A recently published study based on data including our participants identified genes that predispose African American patients to breast cancer. For more information, check out the full study at: https://academic.oup.com/jnci/article/doi/10.1093/jnci/djaa040/5838706.
Permanent link to this article: https://inheritedcancer.net/post80320/
ICARE Social Media Post July 2020
Cancer Risks in Men with BRCA1 and BRCA2 Pathogenic Variants
Check out the article at: https://jamanetwork.com/journals/jamaoncology/article-abstract/2767423
Permanent link to this article: https://inheritedcancer.net/post73120/
ICARE Social Media Post July 2020
Updates to 2020 NCCN Genetic/Familial Colorectal Guidelines
The National Comprehensive Cancer Network (NCCN) released new guidelines for 2020 on July 21, 2020. The big changes included refining some of the risks for genes involved in Lynch Syndrome, and providing specific guidance about cancer screening that may slightly differ by gene. You can check out the full guidelines by creating a FREE account …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post72420/
ICARE Social Media Post July 2020
Racial Inequalities in Genetics
[ngg src=”galleries” ids=”1″ display=”basic_slideshow” arrows=”1″ show_thumbnail_link=”0″]It is important to talk about racial inequalities in healthcare as it affects the care received among Black patients. Through our research efforts, we not only want to address the issue of racism but also think about ways to make healthcare more equitable. A recently published study including our research …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post72120/
ICARE Social Media Post July 2020
Breast Cancer Risks May be Higher in Women with Two CHEK2 Mutations Versus One CHEK2 Mutation
A study comparing women with two CHEK2 mutations to one CHEK2 mutation showed that those with two mutations were: -more likely to get breast cancer (80.6% versus 41.2%) -more likely to be diagnosed at or below age 50 (61.3% versus 23.9%) -more likely to have a second breast cancer diagnosis (22.6% versus 8.1%) These findings …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post71720/
ICARE Social Media Post July 2020
Individuals with a First-Degree Relative with Blood Cancer Are at Increased Risk for Blood-Related Cancer
Individuals with a first-degree relative (for example, parent, sibling, or child) with blood cancer, such as lymphoma, have a higher chance of being diagnosed with blood-related cancer (i.e., hematologic malignancy), according to a recent study. The risk to develop certain blood cancers depends on how closely related the relative is (e.g., higher risks for first-degree …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post71420/
ICARE Social Media Post July 2020
BRCA1/2 and Other Gene Carriers with Breast Cancer Don’t Always Receive Recommended Treatment
BRCA1/2 and other gene mutation carriers with early stage breast cancer are not always receiving cancer treatment as recommended by national guidelines. Even though more and more people have been tested for hereditary cancer over the years, using this information accurately to guide treatment has not been as successful. These findings highlight the need for …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post71020/
ICARE Social Media Post July 2020
Pancreatic Cancer Can Run in Families
Individuals with a family history of pancreatic cancer have a higher risk to develop pancreatic cancer. A recent study reported the following pancreatic cancer risks compared to the general population, based on relationship to an individual with pancreatic cancer: First-degree relative (e.g., parent, sibling, child) – 1.76 fold higher Second-degree relative (e.g., aunts, uncles, grandparents) …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post70720/
ICARE Social Media Post July 2020
Community Spotlight: Mari-Lynn Slayton
In every ICARE Newsletter we feature an ICARE participant as a community spotlight. Our community spotlight in our Winter 2016 ICARE Newsletter is Mari-Lynn Slayton, who was found to have a PALB2 mutation after two breast cancer diagnoses. Check out her story at: https://inheritedcancer.net/community-spotlight/
Permanent link to this article: https://inheritedcancer.net/post70220/
ICARE Social Media Post June 2020
Advances in Treatment for Colorectal Cancer
On June 29, 2020, the FDA approved the use of Keytruda (pembrolizumab), as first-line treatment in unresectable or metastatic, microsatellite instability-high (MSI-H) or mismatch repair-deficient (dMMR) colorectal cancer. Among people with Lynch syndrome, the risk for colorectal cancer (which often has microsatellite instability and/or mismatch-repair deficiency) is raised. Keytruda (pembrolizumab) showed to double the progression-free …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post63020/
ICARE Social Media Post June 2020
Higher Chance of VUS Results in Minorities
Asian, Hispanic, and Black women with breast cancer had higher rates of variants of uncertain significance (VUS) on genetic test results for hereditary cancer genes compared to White women with breast cancer according to a recent study. Check out the full article at https://link.springer.com/article/10.1007%2Fs13187-019-01646-8
Permanent link to this article: https://inheritedcancer.net/post62620/
ICARE Social Media Post June 2020
Community Spotlight: Kelly Frank
June is cancer survivor month! In every ICARE Newsletter we feature an ICARE participant as a community spotlight. Our community spotlight in our Winter 2019 ICARE Newsletter is Kelly Frank, who was found to have a BRIP1 mutation after an ovarian cancer diagnosis. Check out her story at: https://inheritedcancer.net/community-spotlight/
Permanent link to this article: https://inheritedcancer.net/post62320/
ICARE Social Media Post June 2020
Advances in Treatment for BRCA-Mutated Triple Negative Breast Cancer
In a study of 914 women with different breast cancer subtypes, overall pathologic complete response rates were: Higher in those with BRCA1/2 mutations (60.4% versus 46.7%) No differences were seen in those with mutations in other inherited cancer genes Among patients with triple-negative breast cancer, BRCA1/2 mutations had highest response rates to treatment in both …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post61920/
ICARE Social Media Post June 2020
Community Spotlight: Patricia Blumenthal
In every ICARE Newsletter we feature an ICARE participant as a community spotlight. Our community spotlight in our Summer 2018 ICARE Newsletter is Patricia Blumenthal, who was found to have a BRCA2 mutation. Check out her story at: https://inheritedcancer.net/community-spotlight/
Permanent link to this article: https://inheritedcancer.net/post61620/
ICARE Social Media Post June 2020
Advances in BRCA1/2 Breast Cancer Treatment
Through a randomized phase 2 study (called the INFORM trial) among BRCA1/2 carriers with breast cancer, cisplatin was no better in inducing pathologic complete remission compared to AC. The pathologic complete remission rate was 18% for cisplatin and 26% for AC. Cisplatin is not better than other chemotherapy for induction therapy for breast cancers in …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post61220/
ICARE Social Media Post June 2020
Community Spotlight: Christy Mattey
In every ICARE Newsletter we feature an ICARE participant as a community spotlight. Our latest community spotlight in our Winter 2017 ICARE Newsletter is Christy Mattey, who was found to have a CHEK2 mutation after a breast cancer diagnosis. Check out her story at: https://inheritedcancer.net/community-spotlight/
Permanent link to this article: https://inheritedcancer.net/post60920/
ICARE Social Media Post June 2020
Advances in Treatment for Pancreatic Cancer: Cisplatin + Gemcitabine
In BRCA1/2 or PALB2 carriers with stage 3 or 4 pancreatic cancer, the combination of cisplatin + gemtricitabine with veliparib (a PARP inhibitor), did NOT seem to provide additional benefit over cisplatin + gemtricitabine alone. Through this phase 2 randomized control trial, response rates in both treatment arms were high with similar overall survival rates. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post60520/
ICARE Social Media Post June 2020
Community Spotlight: Terry Arnold
June is Cancer Survivor Month! In every ICARE Newsletter we feature an ICARE participant as a community spotlight. Our latest community spotlight in our Summer 2017 ICARE Newsletter is Terry Arnold, who was found to have a BRCA1 mutation after a breast cancer diagnosis. Check out her story at: https://inheritedcancer.net/community-spotlight/
Permanent link to this article: https://inheritedcancer.net/post60220/
ICARE Social Media Post May 2020
Platinum Based Chemotherapy for Metastatic Pancreatic Cancer
A recent study found that patients with metastatic pancreatic cancer who had mutations in the DNA repair genes (either inherited or just in the tumor) had better clinical outcomes after platinum-based chemotherapy compared to patients without these mutations. Check out the link to full article: https://clincancerres.aacrjournals.org/content/early/2020/05/20/1078-0432.CCR-20-0418
Permanent link to this article: https://inheritedcancer.net/post52920/
ICARE Social Media Post May 2020
Cancer Risk Management Among Female BRCA1/2, PALB2, CHEK2, and ATM Carriers in ICARE
A new article was recently published based on data from BRCA1/2, PALB2, CHEK2, and ATM carriers in ICARE. Findings suggest potential overtreatment through risk-reducing surgery among women with pathogenic/likely pathogenic variants in breast cancer genes. This highlights the importance of promoting guideline-adherent, risk-appropriate care. Check out the full article at https://rdcu.be/b4mbg
Permanent link to this article: https://inheritedcancer.net/post52620/
ICARE Social Media Post May 2020
Advances in Treatment for Metastatic Prostate Cancer: Olaparib
On May 19, 2020 the FDA approved the use of olaparib (Lynparza) as treatment in BRCA and other gene carriers (homologous recombination repair genes) with metastatic castration-resistant prostate cancer who have been treated with enzalutamide or abiraterone. Link to full article: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-olaparib-hrr-gene-mutated-metastatic-castration-resistant-prostate-cancer
Permanent link to this article: https://inheritedcancer.net/post52220/
ICARE Social Media Post May 2020
Advances in Treatment for Metastatic Prostate Cancer: Rucaparib
On May 15, 2020 the FDA approved the use of rucaparib (Rubraca) as treatment in BRCA carriers with metastatic castration-resistant prostate cancer who have been treated with androgen receptor-directed therapy and a taxane-based chemotherapy. Link to full article: https://www.fda.gov/drugs/fda-grants-accelerated-approval-rucaparib-brca-mutated-metastatic-castration-resistant-prostate
Permanent link to this article: https://inheritedcancer.net/post51920/
ICARE Social Media Post May 2020
MRI May Detect Breast Cancer Earlier than Mammograms
The National Comprehensive Cancer Network (NCCN) currently recommends an annual breast MRI and an annual mammography for high-risk women (lifetime breast cancer risk at or above 20%). Among women at high risk for breast cancer, MRI detected breast cancer at an earlier stage than mammograms. Women with BRCA1/2 and TP53 were NOT included in this …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post51520/
ICARE Social Media Post May 2020
Advances in Ovarian Cancer Treatment for BRCA1/2 Carriers: Olaparib & bevacizumab
On May 8, 2020 the FDA approved the use of olaparib (Lynparza) as first-line maintenance treatment in BRCA1/2 carriers (deleterious or suspected deleterious mutations) and/or a genomic instability, with advanced epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete or partial response to first-line platinum-based chemotherapy. Link to full article: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-olaparib-plus-bevacizumab-maintenance-treatment-ovarian-fallopian-tube-or-primary
Permanent link to this article: https://inheritedcancer.net/post51220/
ICARE Social Media Post May 2020
Advances in Ovarian Cancer Treatment for BRCA1/2 Carriers: Olaparib
In recognition of World Ovarian Cancer Day, we’d like to share some exciting results from a study of women with ovarian cancer and a BRCA mutation: In a recent phase III trial, olaparib (PARP inhibitor) showed improved response and progression-free survival compared with chemotherapy (without platinum) in BRCA carriers with platinum-sensitive relapsed ovarian cancer who …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post50820/
ICARE Social Media Post May 2020
Advances in Treatment for Metastatic Prostate Cancer: Olaparib
Findings from a recent study showed that olaparib (PARP inhibitor) significantly improved progression-free survival in patients with BRCA1, BRCA2, or ATM genetic alterations. Benefits were also more broadly seen among patients with homologous recombination repair gene defects. Link to full article: https://www.nejm.org/doi/full/10.1056/NEJMoa1911440 Check out the ASCO post article at: https://www.ascopost.com/news/may-2020/olaparib-for-patients-with-mcrpc-and-homologous-recombination-repair-gene-alterations/
Permanent link to this article: https://inheritedcancer.net/post50620/
ICARE Social Media Post May 2020
CDH1: Gastric and Breast Cancer Risks
CDH1 mutation carriers are at higher risk for diffuse gastric cancer (men and women) and lobular breast cancer (women). Current strategies to manage high risks of gastric cancer include prophylactic gastrectomy (i.e., removal of the stomach), which can have severe physical and psychological implications. A recent study found that men have a higher risk of …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post50520/
ICARE Social Media Post May 2020
New Blood Test May Find Ovarian Cancer Early
A study of 80 women identified 4 proteins in blood that may help to detect ovarian cancer early. Further research is still needed with larger groups of women to determine if this new blood test would be a reliable test to find ovarian cancer early. Check out the full article at https://www.nature.com/articles/s41416-019-0544-0
Permanent link to this article: https://inheritedcancer.net/post50120/
ICARE Social Media Post April 2020
Advances in Treatment for Ovarian Cancer in BRCA1/2 Carriers: Niraparib
On April 29, 2020 the FDA approved the use of niraparib (Zeluja) as first-line maintenance treatment in BRCA1/2 carriers with advanced ovarian cancer! More details available at: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-niraparib-first-line-maintenance-advanced-ovarian-cancer
Permanent link to this article: https://inheritedcancer.net/post42920/
ICARE Social Media Post April 2020
PALB2: Cancer Risks and Risk Management
Gene: PALB2 Cancer Risks and Management (per NCCN version 1.2020): Women: Breast cancer risk: Elevated at 53% – Recommend annual breast MRI with contrast starting at age 30, and annual mammogram with consideration of tomosynthesis starting at age 30; Consider risk-reducing mastectomy. Ovarian cancer risk: Elevated at 5% – Manage based on family history. Men …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post42820/
ICARE Social Media Post April 2020
BRCA2: Cancer Risks and Risk Management
Gene: BRCA2 Cancer Risks and Management (per NCCN version 3.2019): Women: Breast cancer risk: Elevated at 60%-70% – Recommend clinical breast exam every 6-12 months starting at age 25, annual breast MRI with contrast starting at age 25, and annual mammogram with consideration of tomosynthesis starting at age 30; consider risk-reducing mastectomy. Ovarian cancer risk: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post42120/
ICARE Social Media Post April 2020
ASCO Guideline: Genetic Testing for Ovarian Cancer
The American Society of Clinical Oncology (ASCO) recently published a guideline reinforcing the longstanding recommendation that all women diagnosed with epithelial ovarian cancer (EOC) be offered genetic testing for hereditary ovarian cancer genes. Many of these women (>15%) have an inherited mutation, most commonly BRCA1 or BRCA2. Identifying BRCA1/2 mutations may help guide cancer treatment. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post41720/
ICARE Social Media Post April 2020
BRCA1: Cancer Risks and Risk Management
Gene: BRCA1 Cancer Risks and Management (per NCCN version 3.2019): Women: Breast cancer risk: Elevated at 60%-70% – Recommend clinical breast exam every 6-12 months starting at age 25, annual breast MRI with contrast starting at age 25, and annual mammogram with consideration of tomosynthesis starting at age 30; consider risk-reducing mastectomy. Ovarian cancer risk: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post41420/
ICARE Social Media Post April 2020
Treatment Patterns in Women with Inherited Breast Cancer
Results from a population-based study of over 20,000 women from SEER registries of Georgia and California diagnosed with stage 0 to stage III breast cancer between 2014 and 2016 tested for inherited breast cancer were recently published. Findings suggest that women who test positive for certain mutations receive certain patterns of treatment. These patterns of …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post41020/
ICARE Social Media Post April 2020
EPCAM: Cancer Risks and Risk Management
Gene: EPCAM Cancer Risks and Management (per NCCN version 3.2019): Women: Endometrial cancer risk: Elevated at 21%-57% – Consider risk-reducing hysterectomy. Ovarian cancer risk: Elevated at 10%-38% – Recommend risk-reducing bilateral salpingo-oophorectomy (removal of ovaries and fallopian tubes). Men and Women: Colorectal cancer risk: Elevated at 43%-52% – Recommend colonoscopy every 1-2 years starting at …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post4120/
ICARE Social Media Post March 2020
Cancer Treatment in Childhood and Treatment-Associated Polyposis
Treatment-associated polyposis (TAP) should be considered in patients who develop many colon polyps after treatment for a childhood or young adulthood cancer but do not have an identified mutation in a hereditary cancer gene. A recent study reported that 35% of patients with TAP developed over 50 colorectal polyps, and 94% had multiple types of …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post32720/
ICARE Social Media Post March 2020
PMS2: Cancer Risks and Risk Management
Gene: PMS2 Cancer Risks and Management (per NCCN version 3.2019): Women: Endometrial cancer risk: Elevated at 0%-15% – Consider risk-reducing hysterectomy. Men and Women: Colorectal cancer risk: Elevated at 12%-20% – Recommend colonoscopy every 1-2 years starting at age 20-25 Gastric cancer risk: Not well established – Consider upper endoscopy every 3-5 years beginning at …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post32420/
ICARE Social Media Post March 2020
Study Based on ICARE Participants with ATM and CHEK2 Mutations
Women with ATM and CHEK2 mutations have a lifetime breast cancer risk greater than 20%, which is the threshold at which screening through a breast MRI is recommended. A recently published study based on ICARE participants with ATM and CHEK2 mutations suggested that most female family members of ATM and CHEK2 mutation carriers do not …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post32020/
ICARE Social Media Post March 2020
MSH6: Cancer Risks and Risk Management
Gene: MSH6 Cancer Risks and Management (per NCCN version 3.2019): Women: Endometrial cancer risk: 17%-46% – Consider risk-reducing hysterectomy. Ovarian cancer risk: 1%-11% – Evidence is insufficient to make specific recommendations. Men and Women: Colorectal cancer risk: 15%-44% – Recommend colonoscopy every 1-2 years starting at age 20-25. Gastric cancer risk: 0%-5% – Consider upper …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post31720/
ICARE Social Media Post March 2020
Polygenic Risk Score and Colon Cancer Risk
A Polygenic Risk Score (PRS) is calculated using genetic differences throughout someone’s DNA, in combination with clinical and family history of cancer. This score, alongside environmental and lifestyle risk factors, may help to identify people who might benefit from screening at an earlier age. Important facts about PRS: – It does NOT look for changes …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post31320/
ICARE Social Media Post March 2020
MSH2: Cancer Risks and Risk Management
Gene: MSH2 Cancer Risks and Management (per NCCN version 3.2019): Women: Endometrial cancer risk: Elevated at 21%-57% – Consider risk-reducing hysterectomy. Ovarian cancer risk: Elevated at 10%-38% – Recommend risk-reducing bilateral salpingo-oophorectomy (removal of ovaries and fallopian tubes). Men and Women: Colorectal cancer risk: Elevated at 43%-52% – Recommend colonoscopy every 1-2 years starting at …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post31020/
ICARE Social Media Post March 2020
Colon Cancer and Polyp Risks
New colon cancer and polyposis genes recently identified include NTHL1, POLE, POLD1, MSH3, and RNF43. Previous studies suggest these genes raise the risk for colon cancer and polyposis; however, recent data suggest that environmental factors may also play an important role in risk. Ultimately, these genes account for a very small fraction of polyposis cases. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post3620/
ICARE Social Media Post March 2020
MLH1: Cancer Risks and Risk Management
Gene: MLH1 Cancer Risks and Management (per NCCN version 3.2019): Women: Endometrial cancer risk: Elevated at 43%-57% – Consider risk-reducing hysterectomy. Ovarian cancer risk: Elevated at 5%-20% – Recommend risk-reducing bilateral salpingo-oophorectomy (removal of ovaries and fallopian tubes). Breast cancer risk: Elevated at 12%-17% – Manage same as general population. Men and Women: Colorectal cancer …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post3320/
ICARE Social Media Post February 2020
Advances in Treatment: Plexiform Neurofibromas in NF1
The FDA has granted a breakthrough therapy designation to selumetinib, a MEK inhibitor, for treatment of inoperable plexiform neurofibromas. These types of neurofibromas are almost exclusively seen in individuals with neurofibromatosis type 1 (NF1). These plexiform neurofibromas are benign tumors on the nerve sheaths and can develop anywhere in the body. These tumors typically cause …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post22820/
ICARE Social Media Post February 2020
Genetic Testing in Tumors versus Genetic Testing for Cancer Risk
There are different types of genetic/DNA tests offered to patients with cancer: 1) Tumor tests, mainly done to guide cancer treatment. 2) Blood or saliva tests (on normal DNA that individual was born with) to identify inherited cancer predisposition, which may also guide cancer treatment in some instances. Genetic testing of the tumor detects mutations …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post22620/
ICARE Newsletter Winter 2020
Community Spotlight
I was diagnosed with breast cancer in December 2018 and was found to be PALB2+. The PALB2 gene had not been tested for when my older sister was diagnosed with breast cancer and had genetic testing done four years earlier. This was new! My cancer was very similar to my sister’s, but being PALB2+ changed …
Continue reading
Permanent link to this article: https://inheritedcancer.net/spotlightnlw2020/
ICARE Newsletter Winter 2020
Treatment Advances Among Those with Inherited Prostate Cancer Predisposition
A recent study reported a high complete response rate among men with a BRCA1/2 mutation with metastatic, castration-resistant prostate cancer who were treated with niraparib (a PARP inhibitor) of 63% compared to 17% in the non-BRCA1/2 group.1 Based on this data, the Federal Drug Administration (FDA) granted breakthrough therapy designation to niraparib on October 3, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nlw2020/
ICARE Newsletter Winter 2020
New Study Based on ICARE Participants with ATM & CHEK2 Mutations
We are excited to tell you about our recently published results based solely on data from ICARE participants with ATM and CHEK2 mutations. Our findings suggest most female family members of ATM and CHEK2 mutation carriers do not qualify for breast MRI screening based on family cancer history alone. This emphasizes the need to share …
Continue reading
Permanent link to this article: https://inheritedcancer.net/icare-newsletter-winter-2020new-study-based-on-icare-participants-with-atm-chek2-mutations/
ICARE Newsletter Winter 2020
Treatment Advances Among Those with Inherited Pancreatic Cancer Predisposition
Results from a recent study showed olaparib (a PARP inhibitor) nearly doubled the progression-free survival in BRCA1/2 carriers with metastatic pancreatic cancer.1 Based on this data, the FDA approved the use of olaparib as a first-line maintenance treatment in BRCA1/2 carriers with metastatic, platinum-sensitive pancreatic cancer. This represents another treatment advance in pancreatic cancer and …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nlw2020/
ICARE Newsletter Winter 2020
PALB2 Mutations & Cancer Risk
A newly published study of 524 families with pathogenic PALB2 mutations from around the world, including almost 50 ICARE participants, represents the largest, most comprehensive effort to evaluate cancer risks.1 Results showed increased risks for female breast cancer (53%), ovarian cancer (5%), pancreatic cancer (2-3%), and male breast cancer (1%). Findings did not suggest higher …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nlw2020/
ICARE Newsletter Winter 2020
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic
There were significant updates and restructuring of the guidelines, with some highlights included below: Substantial reorganization of the guidelines as follows: Now organized by organ site, rather than primarily by certain high penetrance genes Focused efforts to simplify genetic testing criteria Only one flow diagram included, to outline the ‘genetic testing process’ Following scenarios now …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nlw2020/
ICARE Newsletter Winter 2020
Updated Pancreatic Cancer Screening Guidelines through CAPS Consortium
The International Cancer of the Pancreas Screening (CAPS) Consortium recently published updated recommendations about pancreatic cancer screening through MRI/magnetic retrograde cholangiopancreatography (MRCP) and/or an endoscopic ultrasound (EUS).1 Specifically, these guidelines now recommend that individuals with a CDKN2A or STK11 mutation begin screening at age 40. Screening for individuals with a BRCA1/2, ATM, PALB2, MLH1, or …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nlw2020/
ICARE Newsletter Winter 2020
Ask the Expert
Through each newsletter, we give our participants an opportunity to have their questions answered by experts. If you have a question you would like addressed, please email the study team at ICARE@InheritedCancer.net for consideration in future newsletters. The following question was addressed by Ben Ho Park, MD, PhD, who is the Donna S. Hall Chair …
Continue reading
Permanent link to this article: https://inheritedcancer.net/8nlw2020/
ICARE Newsletter Winter 2020
Lynch Syndrome Cancer Risks Across Genes
A worldwide study reporting on cancer risks among individuals with mutations in Lynch syndrome genes showed that there are substantial differences in cancer risks across the various genes.1 Specifically, the risk for colorectal cancer in those with MLH1, MSH2, and MSH6 mutations was substantially higher than what was seen for those with PMS2 mutations. Additionally, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nlw2020/
ICARE Social Media Post February 2020
PMS2 and MSH6 Colorectal Cancer Risks
Individuals with Lynch syndrome have an increased risk of colorectal cancer (CRC) and other cancers. The level of CRC risk is different based on which gene they have a mutation in. Of note, MLH1 and MSH2 carriers have the highest risk of colorectal cancer, generally in the range of 43%-52% by age 70. A recent …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post22020/
ICARE Social Media Post February 2020
Ovarian Cancer Risks in BRCA1/2
The risk of ovarian cancer is raised in women with BRCA1/2 mutations. Recent findings suggest that higher body mass index (BMI) may further raise the risk of ovarian cancer in premenopausal BRCA1/2 carriers. Note that all women with BRCA1/2 mutations are at high risk for ovarian cancer, and should follow current National Comprehensive Cancer Network …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post21820/
ICARE Social Media Post February 2020
Characteristics and Patterns of Spread for BRCA-associated Breast Cancers
BRCA1/2 have distinct characteristics and patterns of spread. A recent study evaluated the characteristics of BRCA1/2 carriers and found that 73% ofBRCA1-associated breast cancers were triple negative; and 72% of BRCA2-associated breast cancers were hormone receptor positive. There were also distinct pattern of spread of breast cancer, with BRCA1 carriers more likely to experience lung …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post21320/
ICARE Social Media Post February 2020
Medication Use for Risk Reduction
Based on studies that show benefits of estrogen and androgen blockers in reducing breast cancer risk in high-risk women, the United States Preventative Service Task Force (USPSTF) recommends that clinicians offer risk-reducing medications to women at increased risk for breast cancer; however, they recommend against routine use of these medications in average-risk women. Hormone receptor …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post21120/
ICARE Social Media Post February 2020
Lynch Syndrome Cancer Risks Across Genes
A worldwide study suggests that risks for cancers for the various Lynch syndrome genes have some differences. The risk of colorectal cancer for those with a mutation in the MLH1, MSH2 and MSH6 genes is higher than what is seen for carriers of a PMS2 mutation. Additionally, men with MSH2 gene mutations have a higher …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post2720/
ICARE Social Media Post February 2020
Differences in Pancreatic Cancer Screening Recommendations from the National Comprehensive Cancer Network (NCCN) and the International Cancer of the Pancreas Screening (CAPS) Consortium
The National Comprehensive Cancer Network (NCCN) and the International Cancer of the Pancreas Screening (CAPS) Consortium recently updated pancreatic cancer screening recommendations. However, there are some differences between these recommendations. Specifically, screening with annual MRI/magnetic retrograde cholangiopancreatography (MRCP) and/or endoscopic ultrasound (EUS) is recommended as follows for NCCN versus CAPS: STK11 regardless of family history: …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post2620/
ICARE Social Media Post February 2020
Updated Pancreatic Cancer Screening Guidelines through the International Cancer of the Pancreas Screening (CAPS) Consortium
The International Cancer of the Pancreas Screening (CAPS) Consortium recently published updated pancreatic cancer screening recommendations. The recommendations include: Screening with MRI/magnetic retrograde cholangiopancreaography (MRCP) and/or endoscopic ultrasound (EUS) The screening was recommended for the following individuals: CDKN2A and STK11 mutation carriers starting at age 40 BRCA1/2, ATM, PALB2, MLH1, and MSH2 mutation carriers (if …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post2420/
ICARE Social Media Post January 2020
Advances in Treatment for Lynch Syndrome-Related Endometrial Cancer
A recent phase II study showed increased response to an immunotherapy drug (avelumab), in women with endometrial cancer with mismatch repair deficiency. Among women with Lynch Syndrome, the risk for endometrial cancer (which often has mismatch repair deficiency) is raised. This new study shows promising treatment options for women with endometrial cancer and Lynch syndrome …
Continue reading
Permanent link to this article: https://inheritedcancer.net/13020/
ICARE Social Media Post January 2020
New Gene: DUOX2
DUOX2 represents a newly identified gene that might predispose patients to thyroid cancer (specifically, ‘non-medullary’ thyroid cancer). The specific gene change (or ‘mutation’) that leads to thyroid cancer predisposition is known as Y1203H Additional studies are needed to confirm this association. Check it out at ASCO Post: https://tinyurl.com/sw3ew63 𝐨𝐫 you can access the article at …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12420/
ICARE Social Media Post January 2020
New Gene: RABL3
About 10% of pancreatic cancers are thought to be the result of an inherited mutation. All of the genes that predispose patients to pancreatic cancers have not yet been discovered. Recently, a new gene known as RABL3 was linked to pancreatic cancer. This gene may increase the risk of hereditary pancreatic cancer. Additional studies are …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12120/
ICARE Social Media Post January 2020
Ovarian Cancer Screening in BRCA1 Carriers
A new study of Polish BRCA1 carriers showed that screening through transvaginal ultrasound was not effective or reliable in detecting ovarian cancer early. Consequently, preventive oophorectomy remains the only proven method to lower ovarian cancer risks and increase survival. Check out the article at: https://www.ncbi.nlm.nih.gov/pubmed/31500890. Check out the NCCN ovarian cancer screening recommendations for BRCA1 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11720/
ICARE Social Media Post January 2020
Racial Disparities in Genetic Testing for Women with Ovarian Cancer
Women of non-European ancestry diagnosed with ovarian cancer have lower rates of referral for genetic testing despite current national guidelines stating ALL women with ovarian cancer and/or a close-blood relative with ovarian cancer should be offered genetic counseling and testing. One study reported that only 1/3 of Black, Latina, and Asian women were referred for …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11620/
ICARE Social Media Post January 2020
Polygenic Risk Scores
A Polygenic Risk Score (PRS) is calculated using genetic differences throughout someone’s DNA, in combination with clinical and family history of cancer. This unique type of score may be utilized to calculate the lifetime risk of breast cancer. Important facts about PRS’s: Different from BRCA1/2 testing, which look for changes in these specific genes Should …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11420/
ICARE Social Media Post January 2020
Women with BRCA1/2: Risk-reducing Mastectomy versus Screening
In a study of 5000 healthy female BRCA1/2 carriers (without a breast cancer diagnosis): BRCA1 carriers: after an average of over 10 years follow-up, survival was higher in those who had bilateral mastectomy (99.7%) compared to those who had breast screening through mammograms and breast MRIs (93%). BRCA2 carriers: no significant differences were seen between …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11020/
ICARE Social Media Post January 2020
Celebrating 10 Years of ICARE
Happy New Year! 2020 represents a decade for ICARE We are celebrating 10 years of research, education, and engagement, through which we have enrolled nearly 3500 participants, including over 2000 gene mutation carriers, disseminated 15 newsletters, led and collaborated on multiple research projects, and impacted individuals affected by inherited cancer predisposition all over the …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post1920/
ICARE Social Media Post January 2020
Advances in Treatment for Pancreatic Cancer in BRCA Carriers
The FDA approved the use of olaparib, a PARP inhibitor, as first-line maintenance treatment in BRCA1/2 carriers with metastatic, platinum-sensitive, pancreatic cancer. Platinum-sensitive cancer is a cancer that responds to treatment with drugs that contain the metal platinum, such as carboplatin or cisplatin. Olaparib showed to nearly double the progression-free survival in BRCA1/2 carriers with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post1320/
ICARE Social Media Post December 2019
Evaluation of PARP Inhibitors in BRCA-Associated Prostate Cancer
The FDA granted breakthrough therapy designation to niraparib (a PARP inhibitor) for the treatment of men with BRCA1/2 positive, metastatic castration-resistant, and heavily pre-treated prostate cancer. Results from a recent study show a 63% complete response rate in men with BRCA1/2 positive, metastatic castration-resistant prostate cancer treated with niraparib compared to 17% in the non- …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post122019/
ICARE Social Media Post December 2019
PALB2 Mutations and Cancer Risk
A new published study of 524 families with PALB2 mutations, including our very own ICARE participants, from around the world showed increased risks of female breast, ovarian, pancreatic, and male breast cancer. The level of risk for female breast cancer is enough to recommend cancer risk management. Cancer risk management includes screening or risk-reducing surgery. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post121619/
ICARE Social Media Post December 2019
American College of Medical Genetics and Genomics: Points-to-Consider Document: “Is there Evidence to Support BRCA1/2 and Other Inherited Breast Cancer Genetic Testing for all Breast Cancer Patients?”
The American College of Medical Genetics and Genomics (ACMG) put forth a Points-to-Consider document: “Is there Evidence to Support BRCA1/2 and Other Inherited Breast Cancer Genetic Testing for all Breast Cancer Patients?” The document discusses: 1) Current data to support testing in breast cancer patients …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post121319/
ICARE Social Media Post December 2019
Patient Reported Outcomes In A Study of PARP Inhibitors in BRCA Carriers with Metastatic Breast Cancer
Did you know? PROs are impacting treatment advances in metastatic breast cancer. Olaparib increased progression-free survival among BRCA carriers with metastatic HER2- breast cancer. Thanks to patient reported outcomes, a new study now suggests it also improved patients’ quality of life! Check it out the new article published in October 2019 directly at https://www.ncbi.nlm.nih.gov/pubmed/31446213!
Permanent link to this article: https://inheritedcancer.net/post12819/
ICARE Social Media Post December 2019
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial Breast, Ovarian, and Pancreatic Guidelines (V1.2020)
We are excited to share the latest version of the NCCN Genetic/Familial Breast, Ovarian and Pancreatic Guidelines (V1.2020), which were just updated. Some of the changes made include: PALB2 was added as a high penetrance gene (similar to BRCA1, BRCA2, CDH1, PTEN and TP53) It is appropriate to consider risk reducing mastectomy for cancer risk management …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post12419/
ICARE Social Media Post December 2019
Explaining Patient Reported Outcomes
The National Quality Forum defines patient reported outcomes (PROs) as “any report of the status of a patient’s health condition that comes directly from the patient.” PROs have the potential to increase overall outcomes and care process and enhance patient and family centered care.
Permanent link to this article: https://inheritedcancer.net/post12319/
ICARE Social Media Post November 2019
Educational Resources: The Family Link Between Breast and Ovarian Cancer in Black Women
The differences seen in access to healthcare across different racial groups are known as health disparities. To address the gap in awareness, we have pursued efforts to raise awareness about inherited breast cancer among African Americans. We are excited to introduce you to another invaluable resource, the Breast Cancer Genetics Research and Education for African American …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post11119/
ICARE Social Media Post November 2019
High Frequency of BRCA in Unselected Women with Metastatic Breast Cancer
Did you know? National practice guidelines currently recommend ALL women with metastatic (HER2-) breast cancer to get genetic testing for inherited cancer (including BRCA1/2 testing), because it can guide eligibility for treatment with PARP inhibitors. A new study led by our colleague at the Vanderbilt-Ingram Cancer Center, Dr. Ben Park, suggests that more women with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post110719/
ICARE Social Media Post October 2019
Lifestyle Factors Associated with Familial and Inherited Breast Cancer Risks
Body mass index (BMI) is an indicator of fat content in the body. A lower body mass index may reduce breast cancer risk in women, including those with a family history of breast cancer. However, a new study reported that higher BMI also increases breast cancer risk in the general population and those with a …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102919/
ICARE Social Media Post October 2019
Physical Activity Associated with Familial and Inherited Breast Cancer Risks
BRCA1/2 carriers may benefit from physical activity to reduce breast cancer risks, just like women in the general population! We know that women in the general population benefit from physical activity to reduce their breast cancer risk, but a recent study showed that this benefit also extends to those with a BRCA1/2 mutation and those with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102819/
ICARE Social Media Post October 2019
BRCA Testing In Black Women
Did you know? Although BRCA testing has been around for over TWO decades, not all populations have benefitted equally from testing. In fact, our previous research has shown that black patients are less aware of BUT interested in genetic testing…when they know about it. In addition, healthcare providers are less likely to identify and suggest …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102419/
ICARE Social Media Post October 2019
Inherited Breast Cancer Among Latina Women
Latina women are at risk for inherited breast cancer: Breast cancer mortality rates are higher among Latina women compared to non-Hispanic White women. Latina women also have a higher prevalence of triple-negative breast cancer when compared to with non-Hispanic White women. Latina women are diagnosed with breast cancer at earlier ages, 56 years versus 63 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post102319/
ICARE Social Media Post October 2019
Advances in Treatment for Advanced Breast Cancer in BRCA Carriers
Monotherapy with PARP inhibitors is FDA-approved for patients with metastatic breast cancer with BRCA mutations. BUT, does adding additional drugs (called ‘combination therapy’) help? In BRCA carriers with metastatic breast cancer, the combination of veliparib AND chemotherapy with platinum-based agents (carboplatin) and taxanes (paclitaxel) led to a longer duration of progression free survival (disease that …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post101819/
ICARE Social Media Post October 2019
Advances in Early Stage Breast Cancer Treatment for BRCA Carriers
New benefits from a PARP inhibitor, talazoparib, among BRCA carriers with early stage breast cancer. The current FDA approvals for the use of PARP inhibitors is limited to women with metastatic (stage IV) breast cancer. These drugs are being tested in early stage breast cancer to shrink down the tumor (called neoadjuvant treatment) before surgery …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post101519/
ICARE Social Media Post October 2019
Male Breast Cancer Risk
Did you know? Beyonce’s father, Matthew Knowles, was diagnosed with breast cancer. He states, “we used to think this was only an issue for women, but this is male or female.” According to CBS news, “he is hoping that sharing his story as man with breast cancer will shine a light on the risk men …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post10619/
ICARE Social Media Post September 2019
Family Sharing Resources: GeneSHARE
With the tremendous advances in gene-based care among those with inherited cancer risk, we are trying to develop tools and strategies to make it easier for more people to benefit from genetic education and testing. We are proud to introduce you to GeneSHARE, a free online toolkit for YOU, to help share positive test results …
Continue reading
Permanent link to this article: https://inheritedcancer.net/post91319/
ICARE Newsletter Summer 2019
Ask the Expert
The following question was addressed by Gillian Hooker, PhD, ScM, LCGC, who is the president-elect for the National Society of Genetic Counselors, Adjunct Associate Professor in the Division of Genetic Medicine at the Vanderbilt University Medical Center, and the Vice President of Clinical Development for Concert Genetics in Nashville, TN. Q. Why was the BRCA1/2 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/10nls2019/
ICARE Newsletter Summer 2019
Updates to National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment: Colorectal Guidelines
(Version 1.2019, posted July 3, 2019) For Individuals with Lynch Syndrome: The cancer risk table was updated: Addition of new cancer risks by specific genes: breast and bladder cancers Updates of cancer risks by specific genes: ovarian, prostate, gastric, pancreatic, urothelial, small bowel, and brain/CNS cancers Removal of reference to sebaceous neoplasms Recommendations for cancer …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nls2019/
ICARE Newsletter Summer 2019
Ovarian Cancer Treatment Advances for BRCA1/2 Carriers
A recently reported study of women with ovarian cancer and homologous recombination deficiency (HRD) who received a PARP inhibitor (niraparib) as fourth line or later treatment showed potential clinical benefit. Specifically, median overall survival after treatment was 19 months in the HRD-positive group (including those with BRCA1/2 mutations) compared to 15.5 months in the HRD-negative …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nls2019/
ICARE Newsletter Summer 2019
New Information About Cancer Risks for Inherited Cancer Genes: BRCA1/2
Looking at pregnancy history and breast cancer risk, a recent study of almost 8,000 women with BRCA1/2 mutations evaluated breast cancer risks related to pregnancy.1 Findings suggested the overall number of pregnancies was not associated with breast cancer risk in BRCA1 carriers; however, BRCA1 carriers with one pregnancy were at higher risk for breast cancer …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nls2019/
ICARE Newsletter Summer 2019
Community Spotlight

Life was great at 45. I had nothing more than a few headaches and was a tad overweight. After a friend was diagnosed with breast cancer, I realized I had not had a mammogram in a couple of years, so I scheduled an appointment. One mass was found, but it was benign and nothing to …
Continue reading
Permanent link to this article: https://inheritedcancer.net/spotlightnls2019/
ICARE Newsletter Summer 2019
Pancreatic Cancer Treatment Advances for BRCA1/2 Carriers
Results from a clinical trial of individuals with a BRCA1/2 mutation and pancreatic cancer showed that patients who received a PARP inhibitor (olaparib) for maintenance treatment had almost half the risk of their disease progressing when compared to receiving a placebo.1 In fact, after 2 years, 22.1% of patients who received olaparib had no disease …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nls2019/
ICARE Newsletter Summer 2019
New Information About Cancer Risks for Inherited Cancer Genes: CHEK2
In a study of inherited mutations in the CHEK2 gene, findings suggest there were two specific mutations that could predispose men to testicular germ cell tumors (TGCT). Specifically, 205 men with these tumors were tested for 48 DNA repair genes, and findings were then tested in other patient populations. These findings suggest that CHEK2 mutations …
Continue reading
Permanent link to this article: https://inheritedcancer.net/7nls2019/
ICARE Newsletter Summer 2019
New Information About Cancer Risks for Inherited Cancer Genes: DICER1
In a study of over 100 individuals with a DICER1 mutation, findings suggest that 5.3% of individuals had developed cancer by age 10, and 19.3% by age 50. After age 10, cancer risks for women were higher compared to men. Specific cancers for which risks were high included gynecologic and thyroid cancers. These findings are …
Continue reading
Permanent link to this article: https://inheritedcancer.net/8nls2019/
ICARE Newsletter Summer 2019
Prostate Cancer Treatment Advances for BRCA1/2 Carriers
There is now information to suggest that identifying inherited mutations in DNA repair genes, such as BRCA1/2 and other genes, in men with metastatic prostate cancer may open doors for other treatment options. Results of a phase 2 clinical trial among men with metastatic and heavily pre-treated prostate cancer were presented at the American Society …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nls2019/
ICARE Newsletter Summer 2019
Expansion of Criteria for BRCA1/2 Testing through the USPSTF
The U.S. Preventive Services Task Force (USPSTF) came out with new genetic testing guidelines for the BRCA1/2 genes, which has garnered substantial media attention. This task force consists of a team of primary care and preventive medicine healthcare experts to lower the chance of a conflict of interest (which is also the reason that subspecialty …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nls2019/
ICARE Newsletter Summer 2019
Expanding Our Thinking About Cancer Risks in TP53 Mutations and Li-Fraumeni Syndrome
Since expanded genetic testing has become available through multigene panel tests, studies have suggested that many people identified to have TP53 mutations do not have a typical personal or family history, which is usually seen with Li-Fraumeni syndrome (LFS). A recent study looking at over 300 individuals with TP53 mutations (identified through multi-gene panel testing) …
Continue reading
Permanent link to this article: https://inheritedcancer.net/11nls2019/
ICARE Newsletter Summer 2019
New Information About Cancer Risks for Inherited Cancer Genes: HAUS6, KANLS1, PCR1
Through a study of over 13,000 patients with serous ovarian cancers and almost 41,000 controls, 34 genes that raise the risk for ovarian cancer were identified. Additional laboratory studies were conducted to further characterize some of these genes, suggesting that three of these new genes may be essential (HAUS6, KANLS1, and PRC1). This study has …
Continue reading
Permanent link to this article: https://inheritedcancer.net/9nls2019/
ICARE Newsletter Winter 2019
Other Advances in Cancer Treatment Among Cancer Patients with Inherited Disease: Familial Adenomatous Polyposis (FAP)
A new drug (sorafenib) showed promising results among patients with desmoid tumors, which are a type of tumor for which patients with Familial Adenomatous Polyposis (FAP) due to APC gene mutations are at risk. These tumors frequently grow and encompass internal organs and can be hard to remove surgically. The newly published research showed that …
Continue reading
Permanent link to this article: https://inheritedcancer.net/9nlw2019/
ICARE Newsletter Winter 2019
Advances in Biomarkers to Detect Pancreatic Cancer Early
Early detection of pancreatic cancer is tremendously important, given that most patients who develop the disease are diagnosed at a later stage of the disease when it is usually incurable. Although screening through imaging studies has been proposed (i.e., magnetic resonance cholangiopancreatography (MRCP) and/or endoscopic ultrasound),1 data to support this type of screening as an …
Continue reading
Permanent link to this article: https://inheritedcancer.net/12nlw2019/
ICARE Newsletter Winter 2019
Community Spotlight

When I was 50 years old I was in pretty good physical shape and I thought I was finally getting six pack abs. I was wrong – those abs were a large football sized tumor, along with a variety of smaller tumors. I was diagnosed with Stage 4 ovarian cancer. I had a hysterectomy and fibroid removal …
Continue reading
Permanent link to this article: https://inheritedcancer.net/spotlightnlw2019/
ICARE Newsletter Winter 2019
Other Advances in Cancer Treatment Among Cancer Patients with Inherited Disease: Lynch Syndrome
Pertaining to metastatic prostate cancer, recently published data reported 8.1% of men with advanced prostate cancer had evidence of mismatch repair (MMR) mutations in their tumors. These types of mutations are frequently seen in tumors among Lynch syndrome patients. In addition, men with this type of tumor had much poorer survival. Tumors with MMR defects …
Continue reading
Permanent link to this article: https://inheritedcancer.net/7nlw2019/
ICARE Newsletter Winter 2019
New Genes: GALNT12
A gene called GALNT12 may be yet another inherited colorectal cancer gene,1 as originally suggested by prior studies.2 The current study screened almost 500 colorectal cancer patients and identified 8 rare variants that may be disease causing. The frequency of variants among colorectal cancer patients was much higher than that observed among population-matched healthy controls, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/13nlw2019/
ICARE Newsletter Winter 2019
Other Advances in Cancer Treatment Among Cancer Patients with Inherited Disease: von Hippel-Lindau (VHL) Disease
Additional exciting advances include the results of a new drug (pazopanib) to treat an inherited cancer condition called von Hippel-Lindau Disease (VHL), in which patients are predisposed to kidney cancers, pancreatic tumors, and hemangioblastomas (i.e., tumors involving the blood vessels). Study results showed that among 31 patients with VHL, overall response rate with the drug …
Continue reading
Permanent link to this article: https://inheritedcancer.net/8nlw2019/
ICARE Newsletter Winter 2019
Basal Cell Cancers May Be a Risk Factor to Predict Inherited Cancer Predisposition
An interesting area of progress to identify individuals with inherited risks included a study of over 13,000 individuals with six or more basal cell cancers (BCC) evaluated through a claims database. Results indicated ~20% of these individuals had a germline mutation in a DNA repair gene, including BRCA1/2, PALB2, and the Lynch syndrome genes, among …
Continue reading
Permanent link to this article: https://inheritedcancer.net/10nlw2019/
ICARE Newsletter Winter 2019
New Research and Approvals of PARP Inhibitor Drugs to Treat Prostate Cancer in BRCA Carriers
Treatment among patients with metastatic castration-resistant prostate cancer: A PARP inhibitor (rucaparib) was granted a breakthrough therapy designation in October 2018 for monotherapy (i.e., sole treatment) among men with metastatic castration-resistant prostate cancer (with a BRCA1/2 mutation) who have received at least one prior androgen receptor-directed treatment and taxane-based chemotherapy. This designation was granted based …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nlw2019/
ICARE Newsletter Winter 2019
Ask the Expert
The following question was addressed by Georgia Wiesner, MD, MS, a nationally renowned clinical cancer geneticist, who is an Ingram Professor of Cancer Research, Professor of Medicine in the Division of Genetic Medicine, and the Director of the Clinical and Translational Hereditary Cancer Program for the Vanderbilt-Ingram Cancer Center in Nashville, Tennessee. Q. What are …
Continue reading
Permanent link to this article: https://inheritedcancer.net/14nlw2019/
ICARE Newsletter Winter 2019
New Research and Approvals of PARP Inhibitor Drugs to Treat Breast Cancer in BRCA Carriers
Treatment among patients with advanced or metastatic breast cancer: A PARP inhibitor (talazoparib) was approved by the FDA on October 16, 2018 for BRCA carriers with HER2-negative locally advanced or metastatic breast cancer, based on results of the EMBRACA trial outlined in the last ICARE newsletter. Litton JK, et al. N Engl J Med. 2018 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nlw2019/
ICARE Newsletter Winter 2019
Expansion of Lynch Syndrome Tumor Spectrum Which May Have Treatment Implications
Although the Lynch syndrome tumor spectrum is thought to be limited to cancers of the colorectum, endometrium, ovaries, stomach, and a few other cancer types, a recent article suggested there might be a broader tumor spectrum than previously considered. Furthermore, colorectal and endometrial cancers which develop among Lynch syndrome patients frequently are determined on tumor …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nlw2019/
ICARE Newsletter Winter 2019
Refining Treatment for Aggressive Prostate Cancer in Men with BRCA2
A recent study reported that a large proportion of men with aggressive prostate cancer have inherited cancer gene mutations. Specifically, among 400 patients with castration-resistant prostate cancer, 16.2% had a germline mutation in a DNA damage repair gene, including 3% with a BRCA2 mutation. Among BRCA2 carriers, survival was 17.4 months, which was much lower …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nlw2019/
ICARE Newsletter Winter 2019
New Research and Approvals of PARP Inhibitor Drugs to Treat Ovarian Cancer in BRCA Carriers
First line maintenance treatment among patients newly diagnosed with advanced ovarian cancer: The results of a trial using a PARP inhibitor (olaparib) as maintenance treatment among ovarian cancer patients with advanced disease, a BRCA mutation, and complete or partial response to platinum-based chemotherapy showed that survival at 3 years was 60% among those who got …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nlw2019/
ICARE Newsletter Winter 2019
High Proportion of Inherited Genes Detected Among Patients with Advanced Renal Cancer
In a study of advanced renal cell cancer patients, inherited cancer gene mutations were identified in 16%, of which 5.5% had mutations in genes related to renal cell cancer. Among the subgroup with non-clear cell renal cancer, about a fifth had an inherited cancer gene mutation, half of which had a gene mutation that could …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nlw2019/
ICARE Newsletter Winter 2019
Testing Interpretation and Variant Reclassification
Results of germline genetic testing generally yield three types of test results: Deleterious (positive), Negative (no mutation detected), and Variant of Uncertain Significance (VUS). As more genes are tested, the chance for a positive result goes up, as does the chance of receiving a VUS result.1 VUS results tell us that it remains uncertain whether …
Continue reading
Permanent link to this article: https://inheritedcancer.net/15nlw2019/
ICARE Newsletter Winter 2019
New Online Risk Calculator to More Accurately Predict Breast Cancer Risk
Prediction of breast cancer risk is important to identify those at highest and lowest risks, to help guide screening. A previously developed risk algorithm called Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm (BOADICEA) was recently extended to include truncating mutations in the BRCA genes, PALB2, CHEK2, and ATM. This online risk …
Continue reading
Permanent link to this article: https://inheritedcancer.net/11nlw2019/
ICARE Newsletter Summer 2018
The Role of Inherited Genes Increasingly Recognized in Pancreatic Cancer
A number of recent studies have suggested that a substantial number of individuals with pancreatic cancer have a mutation in an inherited cancer gene. In a study of over 300 patients with pancreatic cancer (and with one or two family members with pancreatic cancer), 12% were found to have a mutation in 1 of 11 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nls2018/
ICARE Newsletter Summer 2018
Community Spotlight

I was aware from a very young age that breast cancer was part of our family. I knew that my great-grandmother (whom I never met) had breast cancer and my grandmother was diagnosed in her 50’s. While I didn’t grow up being afraid of the disease, I was far more aware of it than were …
Continue reading
Permanent link to this article: https://inheritedcancer.net/spotlightnls2018/
ICARE Newsletter Summer 2018
Updates to NCCN Genetic/Familial High-Risk Assessment: Breast and Ovarian Guidelines
(Version 1.2019, posted July 11, 2018) Regardless of family history, some individuals with a hereditary breast- and ovarian-related cancer may benefit from genetic testing to determine eligibility for targeted treatment The multi-gene testing section table was updated with: A potential association of ATM with ovarian cancer risk Potential increased risk of BARD1 with breast cancer …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nls2018/
ICARE Newsletter Summer 2018
Prevention of Colorectal Cancer Among Individuals with Familial Adenomatous Polyposis (FAP)
Through a randomized trial, patients with FAP were treated with sulindac and erlotinib versus placebo for 6 months. Results of the study showed that those treated with sulindac and erlotinib had 70% fewer polyps than those in the placebo group. The lower number of polyps was seen in both those with an intact colorectum and …
Continue reading
Permanent link to this article: https://inheritedcancer.net/9nls2018/
ICARE Newsletter Summer 2018
NTHL1: A New Gene for Inherited Colorectal Cancers
In a study of 51 individuals with multiple colon polyps drawn from 48 families, genetic testing through whole-exome sequencing identified 7 individuals (from 3 unrelated families) to have a mutation in both copies of their NTHL1 gene, and pedigree structure was consistent with autosomal recessive inheritance.1 All these individuals had colorectal cancer and a large …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nls2018/
ICARE Newsletter Summer 2018
Updates to NCCN Genetic/Familial High-Risk Assessment: Colorectal Guidelines
For Individuals with Lynch Syndrome: Surveillance for gastric and small bowel cancer now indicates there is no clear data to support this, but surveillance can be performed every 3-5 years starting at age 40 Lack of evidence to make a recommendation for pancreatic or prostate cancer screening, beyond those already recommended through other NCCN Guideline …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nls2018/
ICARE Newsletter Summer 2018
Another PARP-Inhibitor Trial Among BRCA Carriers with Advanced Breast Cancer
In a Phase 3 clinical trial among BRCA carriers with advanced breast cancer, an oral PARP Inhibitor (talazoparib) was compared to standard chemotherapy. Among those who received the PARP inhibitor, risk of disease progression or death was 46% lower, and the response rate was double. Furthermore, the side effect profile, quality-of-life measures, and breast cancer …
Continue reading
Permanent link to this article: https://inheritedcancer.net/8nls2018/
ICARE Newsletter Summer 2018
Inherited Leukemias: The Importance of TP53/Li-Fraumeni Syndrome and Other Genes
It has long been established that the risk for developing leukemia in childhood is high among individuals with Li-Fraumeni Syndrome; however, better understanding the characteristics of leukemia among these individuals is important to guide treatment approaches. In a study of children with Acute Lymphocytic Leukemia (ALL), those with a germline TP53 mutation (compared to those …
Continue reading
Permanent link to this article: https://inheritedcancer.net/11nls2018/
ICARE Newsletter Summer 2018
Differences in Breast Cancer Risks Among Women with Lynch Syndrome
Breast cancer risks were recently reported among a sample of 423 women with mutations in one of the Lynch syndrome genes (MLH1, MSH2, MSH6, or PMS2).1 Results indicated that breast cancer risks were substantially higher among those with MSH6 and PMS2 mutations, compared to MLH1 and MSH2 mutations. In fact, breast cancer risk to age …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nls2018/
ICARE Newsletter Summer 2018
New Data to Suggest Additional Genes Associated with Breast and Ovarian Cancer
A recent study reported on cancer risks among over 10,000 cancer patients across the United States who had genetic testing. Findings suggest breast cancer risks were associated with ATM, CHEK2, and PALB2, as expected; but an association was also found with MSH6 (in line with other recently published data, as outlined in another article in …
Continue reading
Permanent link to this article: https://inheritedcancer.net/7nls2018/
ICARE Newsletter Summer 2018
Refining Cancer Risks Among Individuals with Lynch Syndrome
Over the past year, multiple studies have refined risks and types of cancer among individuals with Lynch syndrome. Through a Scandinavian study, risks for 13 types of cancer (with colorectal cancers being excluded), were reported to be elevated with differences related to gender, age, and the gene in which mutation was present. Incidence rates of …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nls2018/
ICARE Newsletter Summer 2018
Ask the Expert
The following question was addressed by Ronald D. Alvarez, MD, MBA who is Professor, Chairman, and Clinical Service Chief of the Department of Obstetrics and Gynecology at Vanderbilt University Medical Center in Nashville, Tennessee. Dr. Alvarez has been the recipient of several National Cancer Institute (NCI) and other industry funded grants in support of his …
Continue reading
Permanent link to this article: https://inheritedcancer.net/10nls2018/
ICARE Newsletter Winter 2018
Community Spotlight

A cancer diagnosis is a life-changing event for every patient and their extended family. However, how we respond to this diagnosis are as individual as our very existence as evidenced by our looks and personalities. Following my diagnosis of stage 4 prostate cancer in 2014 at age 54 which had spread to my bones, I …
Continue reading
Permanent link to this article: https://inheritedcancer.net/spotlightnlw2018/
ICARE Newsletter Winter 2018
Advances in the Understanding of Inherited Prostate Cancer
Findings through a recent study reported that inherited cancer gene mutations were present in 8.2% of those with advanced or metastatic prostate cancer, which provides additional support to include this group of men in broader testing, particularly as targeted treatments based on inherited gene mutations becomes increasingly available.1 Another recent study suggested that those with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/10nlw2018/
ICARE Newsletter Winter 2018
The Role of “Non-Truncating” Mutations in RAD51D on Ovarian Cancer Risk
As testing has broadened to include newer inherited cancer genes, studies have suggested that mutations which shorten the protein (“truncating mutations”) in the RAD51D gene are associated with ovarian cancer. However, a recent study examined ovarian cancer risks for a “non-truncating” change (a single base pair within the gene is changed which is called a …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nlw2018/
ICARE Newsletter Winter 2018
Updates to NCCN Genetic/Familial High-Risk Assessment: Breast and Ovarian Guidelines
(Version 1.2018, posted Oct. 3, 2017) Metastatic prostate cancer was added as an indication for evaluation and testing for the BRCA1 and BRCA2 genes Among BRCA1, BRCA2, TP53 and PTEN carriers, women between ages 25-29 may consider having an annual mammogram with consideration of tomosynthesis if a breast MRI is not available. Among female BRCA2 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nlw2018/
ICARE Newsletter Winter 2018
Getting Closer to Detecting Cancers Early Through a Blood Test?
A recent study reported on a single blood test, named “CancerSEEK”, which can screen for 8 common types of cancer (ovarian, liver, stomach, pancreas, esophagus, colorectal, lung, and breast) and may help to identify the location at which the cancer started. This test evaluated the blood from over 1000 patients with Stage 1 to 3 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nlw2018/
ICARE Newsletter Winter 2018
Advances in New Treatments for Individuals with Lynch Syndrome
A recently published phase II clinical trial investigated the use of a new class of drugs (called PD-1 Inhibitors) in DNA mismatch repair-deficient/ microsatellite instability-high colorectal tumors (which are features seen in the majority of colorectal tumors from individuals with Lynch Syndrome) among patients with metastatic disease.1 Investigators found patients who received two PD-1 Inhibitors …
Continue reading
Permanent link to this article: https://inheritedcancer.net/7nlw2018/
ICARE Newsletter Winter 2018
Refining Risks and Outcomes of Breast Cancer in BRCA Carriers
In an effort to further study breast cancer risks among BRCA carriers, a recently published study compared breast cancer risks among those with and without a close family member (first-degree relative) with breast cancer.1 Findings showed that risk for breast cancer by age 80 was 60.8% in BRCA1 carriers and 63.1% among BRCA2 carriers, with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nlw2018/
ICARE Newsletter Winter 2018
FDA Approval of PARP Inhibitor (Lynparza) for Treatment of Advanced Breast Cancer
On January 12, 2018, the FDA approved the first PARP Inhibitor (Lynparza) for treatment in patients with advanced breast cancer due to inherited BRCA mutations.1 This drug is already approved for certain BRCA carriers for advanced ovarian cancer. PARP inhibitors were originally developed to target the specific pathway through which cancer develops among those with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nlw2018/
ICARE Newsletter Winter 2018
Ask the Expert
The following question was addressed by Dr. Ingrid Meszoely is a breast surgeon and Clinical Director of the Vanderbilt Breast Center at One Hundred Oaks. She leads a high-risk clinic, through which she and her team of nurse practitioners manage patients with inherited breast cancer predisposition. Her research interests include both clinical and translational breast …
Continue reading
Permanent link to this article: https://inheritedcancer.net/8nlw2018/
ICARE Newsletter Winter 2018
Study Suggests Inherited Cancer Genes Are Important in Pancreatic Cancer
In a recent study which included over 800 patients with pancreatic ductal cancer, inherited cancer gene mutations were found in a much higher proportion than expected. Almost 5% of these patients had mutations identified in inherited cancer genes, the majority of which were in genes thought to be associated with pancreatic cancer (including BRCA2, ATM, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/9nlw2018/
ICARE Newsletter Winter 2018
Advances in Cancer Screening Among Li-Fraumeni Syndrome Patients
Several research groups from around the world that have conducted cancer screening among patients with Li-Fraumeni syndrome and a germline TP53 mutation have recently reported on their observations. Specifically, the National Cancer Institute group demonstrated that screening inclusive of rapid total body MRI detected cancers at an early stage,1 similar to findings published through other …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nlw2018/
ICARE Newsletter Summer 2017
Ask the Expert
The following question was addressed by Dr. Steven Narod who is a Tier I Canada Research Chair in Breast Cancer and a senior scientist at the Women’s College Research Institute in Toronto, Canada. Dr. Narod is a world-leader in the field of breast and ovarian cancer genetics. Q. In women with a BRCA mutation and …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nls2017/
ICARE Newsletter Summer 2017
Emerging FDA Approvals of Immunotherapy Among Patients With Metastatic MSI-H Cancers
Over the last few years, immunotherapy has emerged as an exciting new class of drugs. As early as 2015, immunotherapy through PD-1 Inhibitors among patients with MSI-H colorectal cancers was shown to be of potential benefit.1 As many individuals with Lynch Syndrome have cancers that are MSI-H and mismatch repair deficient, this class of drugs …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nls2017/
ICARE Newsletter Summer 2017
Breast and Ovarian Cancer Risks Among BRCA Carriers Followed Over Time
Findings from an international study of over 6000 women with a BRCA1 mutation and almost 4000 women with a BRCA2 mutation followed for an average of 5 years were recently published.1 Results showed the risk of breast cancer by age 80 was ~70% for both BRCA1 and BRCA2 carriers. Rates of breast cancer increased until …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nls2017/
ICARE Newsletter Summer 2017
Community Spotlight

After a long rainy summer filled with doctor visits, I was finally diagnosed with triple-negative inflammatory breast cancer (TN IBC) at the age of 49. I completed treatment in June 2008 and was grateful to have a new phase to my vocabulary – NED, ‘no evidence of disease’. Since there was no cancer history in my …
Continue reading
Permanent link to this article: https://inheritedcancer.net/spotlightnls2017/
ICARE Newsletter Summer 2017
New Results of a PARP Inhibitor Study Among BRCA Carriers with Metastatic Breast Cancer
Over the last decade, a new class of drugs called “PARP Inhibitors” has been evaluated as a form of targeted treatment among BRCA carriers. Results were recently reported from a Phase 3 clinical trial among BRCA carriers with HER2-negative metastatic breast cancer who received two or less prior chemotherapy regimens for their metastatic disease. Study …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nls2017/
ICARE Newsletter Summer 2017
What Are the Benefits of Adding a Mammogram to MRI for Breast Cancer Screening Among Women with BRCA Mutations?
Recently, researchers evaluated the benefit of adding a mammogram to MRI for breast cancer screening among ~2000 women with a BRCA1 or BRCA2 mutation. Results indicated that the addition of mammography to MRI did not substantially raise the chance of detecting breast cancer in the overall group. However, one-third of breast cancer cases diagnosed among …
Continue reading
Permanent link to this article: https://inheritedcancer.net/7nls2017/
ICARE Newsletter Summer 2017
What Are New and Subsequent Cancer Risks Among Patients with Li-Fraumeni Syndrome?
Although individuals with Li-Fraumeni Syndrome (LFS), due to mutations in the TP53 gene, have a very high lifetime risk of cancer, risks of initial and subsequent cancers are not well defined. Through a group of patients with the classic form of LFS, researchers at the National Cancer Institute estimated their cancer risks. They evaluated a …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nls2017/
ICARE Newsletter Summer 2017
Breast and Ovarian Cancer Associations for Genes Tested Through Multi-Gene Panels
As testing for multiple genes at the same time (“multi-gene panel testing”) has become increasingly available with tremendous advances in genetic testing technology, it has become critical to evaluate and refine cancer associations and levels of risk for many of these genes now tested. Through a commercial laboratory database of almost 100,000 results of multi-gene …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nls2017/
ICARE Newsletter Winter 2017
Community Spotlight

On my 43rd birthday I was diagnosed with an advanced stage breast cancer. Although my BRCA1 and BRCA2 results were surprisingly negative, I was certain there must be a genetic component to my breast cancer since I was diagnosed at a fairly young age. I remained in contact with my geneticist, Dr. Georgia Wiesner, and …
Continue reading
Permanent link to this article: https://inheritedcancer.net/spotlightnlw2017/
ICARE Newsletter Winter 2017
Newly Approved PARP-Inhibitor (Rucaparib) to Treat BRCA Carriers with Ovarian Cancer
The FDA just approved another PARP inhibitor, rucaparib, for BRCA carriers with ovarian cancer who have already been treated with two or more chemotherapies. Among those with BRCA-mutant ovarian cancers, 54% had a partial or complete response to the drug with a median duration response of 9.2 months. The agency also approved a companion diagnostic …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nlw2017/
ICARE Newsletter Winter 2017
Characterizing Breast Cancers That Develop Among Women with a CHEK2 Mutation
With increasing use of multi-gene panel tests, one of the genes in which mutations are frequently detected among breast cancer patients and others is the CHEK2 gene. This gene has been shown to have a 2-3 fold excess risk for breast cancer. There are many CHEK2 mutations that have been identified that generally fall into …
Continue reading
Permanent link to this article: https://inheritedcancer.net/8nlw2017/
ICARE Newsletter Winter 2017
New Study Suggesting BRCA1/2 and ATM Are Associated with Aggressive Prostate Cancer
Among 799 patients with prostate cancer, the rate of BRCA1/2 mutations was much higher among those who passed away of prostate cancer (6.07%) compared to those with low risk disease (1.44%).1 Among the group that died of prostate cancer, those with BRCA1/2 or ATM mutations passed away at an earlier age and had a shorter …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nlw2017/
ICARE Newsletter Winter 2017
NCCN Guidelines Version 1.2017: Genetic/Familial High-Risk Assessment: Breast and Ovarian
Additional guidance pertaining to cancer risk management was provided in the most recent version of the NCCN Guidelines for inherited breast and ovarian cancer. These guidelines now include an expanded table outlining cancer risks and management for each gene, taking into account the age at initiation of each risk management modality as well as footnotes …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nlw2017/
ICARE Newsletter Winter 2017
A Newly Identified Inherited Colon Cancer Gene: FAN1
There continues to be rapid advances in identifying new genes involved in inherited cancer risk. An example of yet another recently identified gene is FAN1, in which a nonsense variant (i.e. the premature change or loss of a protein) was identified following exome sequencing in 3 individuals from a family who met clinical criteria for …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nlw2017/
ICARE Newsletter Winter 2017
Ask the Expert
The following question was addressed by Dr. Steven Narod who is a Tier I Canada Research Chair in Breast Cancer and a senior scientist at the Women’s College Research Institute in Toronto, Canada. Dr. Narod is a world-leader in the field of breast and ovarian cancer genetics. Q. Does salpingo-oophorectomy reduce the risk of breast …
Continue reading
Permanent link to this article: https://inheritedcancer.net/7nlw2017/
ICARE Newsletter Winter 2017
The Potential Promise of Immunotherapy Targeted to Those with Bi-Allelic Mutations in Lynch Syndrome Genes
People with Lynch Syndrome have a non-working Lynch gene (“mutation”), while the other copy of that gene is normal (recognizing that all of these genes come in pairs, with one member of the pair coming from each parent). Over the last few years, there has been an increased realization that some individuals have a mutation …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nlw2017/
ICARE Newsletter Winter 2017
How Does Having a Mother with Breast Cancer and a BRCA Mutation Affect Adolescent Girls?
A recent study compared psychosocial adjustment and risk perception among 11 to 19 year old daughters of women with breast cancer, comparing those with a BRCA mutation versus those without.1 The overall findings from the study were reassuring, suggesting that adolescent girls from BRCA-positive families had higher self-esteem and similar psychosocial adjustment compared to their …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nlw2017/
ICARE Newsletter Summer 2016
Cancer Chemoprevention in Individuals with Familial Adenomatous Polyposis (FAP)
Prior research has demonstrated that NSAIDs significantly reduce colonic and rectal polyp burden among individuals with FAP although their impact on outcomes remains to be determined.1,2 Recent data extended these results to the small intestine through completion of a randomized clinical trial among patients with FAP which demonstrated that use of sulindac and erlotinib compared …
Continue reading
Permanent link to this article: https://inheritedcancer.net/7nls2016/
ICARE Newsletter Summer 2016
Practice Guideline Updates for NCCN Genetic/Familial High-Risk Assessment
The National Comprehensive Cancer Network (NCCN) is a network of oncology healthcare providers who work together to develop best practice guidelines for the delivery of cancer care. Given the increasing use of testing for mutations in several inherited cancer genes at one time (called “multi-gene panel testing”), the Breast/Ovarian and Colorectal Panels sought to provide …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nls2016/
ICARE Newsletter Summer 2016
What Are the Endometrial Cancer Risks Among BRCA Carriers?
Although BRCA mutations confer increased risk for ovarian, fallopian tube, and primary peritoneal cancer, there have been limited and conflicting risks reported for endometrial cancer. Consequently, current practice guidelines only recommend the removal of the fallopian tubes and ovaries as a risk-reducing option for BRCA carriers.1 Specifically, through a prospective study of 4500 women with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/9nls2016/
ICARE Newsletter Summer 2016
CHEK2 *1100delC Mutation Carriers: Breast Cancer Risk by Age and Tumor Type and Other Associated Cancer Risks
The CHEK2 *1100delC mutation is the most common “truncating” mutation (causing a shortened protein) in the CHEK2 gene among Europeans, with lifetime breast cancer risk in the range of 20-30% among female carriers. Results of data pooled from over 30 studies which included 40,000 breast cancer cases and 40,000 controls, showed that estrogen receptor (ER) …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nls2016/
ICARE Newsletter Summer 2016
Practice Guideline Updates for NCCN Genetic/Familial High-Risk Assessment
The National Comprehensive Cancer Network (NCCN) is a network of oncology healthcare providers who work together to develop best practice guidelines for the delivery of cancer care. Given the increasing use of testing for mutations in several inherited cancer genes at one time (called “multi-gene panel testing”), the Breast/Ovarian and Colorectal Panels sought to provide …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nls2016/
ICARE Newsletter Summer 2016
Risk of Second Cancers Among Those with PTEN Mutations
A recently published study to evaluate the risk of second cancers among PTEN mutation carriers showed that women with breast cancer had a 10-year second breast cancer cumulative risk of almost 30%. Overall, the risk of second primary cancers was almost 8-fold that of the general population, primarily due to the higher risks of cancer …
Continue reading
Permanent link to this article: https://inheritedcancer.net/8nls2016/
ICARE Newsletter Summer 2016
Ask the Expert
The following question was addressed by Dr. Christine Laronga at the Moffitt Cancer Center: Q. How should bone health be monitored in women with a BRCA mutation after removal of the ovaries (i.e., risk-reducing salpingo-oophorectomy (RRSO))? A. Women with a BRCA mutation have a substantially high risk to develop ovarian cancer in their lifetime, yet …
Continue reading
Permanent link to this article: https://inheritedcancer.net/10nls2016/
ICARE Newsletter Summer 2016
Surveillance Among Individuals with Li-Fraumeni Syndrome (LFS): An 11 Year Follow-Up Study
Results from the original screening protocol for LFS1 were recently updated following collection of 11 years of follow-up data.2 Through this study, 89 patients with LFS were given the option of a clinical surveillance protocol consisting of a physical examination as well as frequent biochemical and imaging studies. Forty asymptomatic tumors were detected in 32% …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nls2016/
ICARE Newsletter Summer 2016
Inherited Cancer Genes and Metastatic Prostate Cancer
Several prior studies have suggested that men with a BRCA mutation (primarily BRCA2) tend to develop an aggressive form of prostate cancer that is more likely to metastasize. These findings were recently extended through a new study published in the New England Journal of Medicine.1 In this study, almost 700 men with metastatic prostate cancer, …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nls2016/
ICARE Newsletter Summer 2016
Community Spotlight

When I was diagnosed with cancer the first time at age 38, my sister (a breast cancer survivor since the age of 29) was positive we had a BRCA gene mutation. However, after we both had genetic testing done in 2006 the results showed we didn’t. Doctors said they were surprised we did not have a mutation in one of …
Continue reading
Permanent link to this article: https://inheritedcancer.net/spotlightnls2016/
ICARE Newsletter Summer 2016
An Approach to Making Risk Management Recommendations for Newer Inherited Cancer Genes
A recent article sought to develop an approach to cancer risk management among individuals with mutations in newer inherited cancer genes, many of which result in a moderate (rather than ‘high’) cancer risk. Overall, the investigators suggest a framework that takes the age-specific, lifetime, and absolute cancer risks into account for inherited cancer genes where …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nls2016/
ICARE Newsletter Winter 2016
Improving Our Understanding of Cancer Risks Among Individuals with Li-Fraumeni Syndrome
A recent study from France included over 400 patients with Li-Fraumeni Syndrome (all of whom had an inherited TP53 gene mutation). Cancer types among children and adults differed, with the main cancer types among children being osteosarcomas, adrenocortical carcinomas, central nervous system (CNS) tumors and soft tissue sarcomas; whereas among adults, the main cancer types …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nlw2016/
ICARE Newsletter Winter 2016
How Much Does Age at First Breast Cancer Affect the Risk of Contralateral Breast Cancer Risk in BRCA Carriers?
A recently published study of Dutch patients (including 200 BRCA1 carriers, 71 BRCA2 carriers, and 6023 non-carriers) showed that the contralateral breast cancer (breast cancer in the opposite breast) risks at 10 years was 21.1% for BRCA1, 10.8% for BRCA2, and 5.1% for non-carriers. Among BRCA mutation carriers, it was important to take age at …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nlw2016/
ICARE Newsletter Winter 2016
Ask the Expert
The following question was addressed by Dr. Christine Laronga at the Moffitt Cancer Center: Q. As a BRCA carrier, is it reasonable for me to consider nipple-sparing mastectomy (compared to total mastectomy) to reduce my future risks of breast cancer? A. One strategy to manage the high (60-70%) lifetime breast cancer risk among women with …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nlw2016/
ICARE Newsletter Winter 2016
Community Spotlight
I was first diagnosed with breast cancer when I was 56 years old. Because of my strong family history of breast cancer, I was referred for genetic counseling and had BRCA testing at that time. Recently, I was diagnosed with breast cancer again. When I went to see my surgeon, she advised me to have more genetic …
Continue reading
Permanent link to this article: https://inheritedcancer.net/spotlightnlw2016/
ICARE Newsletter Winter 2016
Potential Use of PARP-Inhibitors Among Men with Prostate Cancer Who Carry a Mutation in BRCA or Other DNA-Repair Gene
A recent study published in the New England Journal of Medicine suggests that PARP-Inhibitors may be of potential use in men who are no longer responding to standard treatments and carry either somatic (i.e., tumor) and/or germline (inherited) mutations in DNA-repair genes (i.e., BRCA1/2, ATM, Fanconi Anemia genes and CHEK2).1 Of 49 men with prostate …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nlw2016/
ICARE Newsletter Winter 2016
The Importance of Sharing Genetic Test Results with Family Members
Once an individual has had genetic testing for inherited cancer predisposition this information could help their close family members. For example, when a BRCA mutation or a mutation in another inherited cancer gene is found, it is important for close family members (with or without a diagnosis of cancer) to know so they too can …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nlw2016/
ICARE Newsletter Winter 2016
What Is the Risk for Ovarian Cancer Among Women with Mutations in Newer Ovarian Cancer Genes?
The most common form of inherited ovarian cancer is due to mutations in the BRCA1 and BRCA2 genes, which are present in 10-15% of women with ovarian cancer and lead to an ovarian cancer risk of up to 44% and 27%, respectively. Another set of genes known to raise ovarian cancer risks are the mismatch …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nlw2016/
ICARE Newsletter Winter 2016
More Information About the Inherited Component of Pancreatic Cancer
Although pancreatic cancer is one of the cancer types seen among individuals with mutations in inherited cancer genes (including BRCA2 and BRCA1), the proportion of individuals with pancreatic cancer who have an inherited cause has remained uncertain. To further clarify the role of BRCA1 and BRCA2 (BRCA), over 300 patients with pancreatic cancer were tested …
Continue reading
Permanent link to this article: https://inheritedcancer.net/7nlw2016/
ICARE Newsletter Summer 2015
The Rapid Pace of Discovering More Inherited Cancer Genes Continues
Over the last few months, a number of additional genes associated with inherited cancer predisposition have been identified. A few of these genes include: 1) the RECQL gene which appears to be another rare gene involved in inherited breast cancer1; 2) the SMAD9 gene associated with hamartomatous polyposis and ganglioneuromas of the intestinal tract2; 3) …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nls2015/
ICARE Newsletter Summer 2015
Location and Type of BRCA1/2 Mutation May Impact Breast and Ovarian Cancer Risks
A study of almost 20,000 BRCA1 carriers and 12,000 BRCA2 carriers demonstrated differences in breast and ovarian cancer risks depending on the location and type of mutation. Although all regions are associated with increased risk for breast and ovarian cancers among BRCA1/2 carriers, there were specific regions that were associated with even higher cancer risks. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nls2015/
ICARE Newsletter Summer 2015
Advances in Preventive and Treatment Approaches for Individuals with Lynch Syndrome
A study of over 1800 individuals with a mutation in one of the Lynch Syndrome genes was recently completed to assess whether aspirin and ibuprofen use may reduce colon cancer risk. Results showed that in those who took aspirin or ibuprofen for between 1 month and 4.9 years, the colon cancer risks were lower than …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nls2015/
ICARE Newsletter Summer 2015
Breast Cancer Risks and Outcomes Among Women with a PALB2 Mutation
Through a recent study of over 12,000 Polish women with breast cancer, PALB2 mutations were detected in almost 1%. In this study, about one third of those with a PALB2 mutation had triple negative (lacking estrogen, progesterone and HER2 receptors) breast cancer and the average age at breast cancer diagnosis was 53.3 years. Breast tumors …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nls2015/
ICARE Newsletter Summer 2015
2015 NCCN Clinical Practice Guideline Update
Breast and Ovarian Management Based on Genetic Test Resultsa Recommend Breast MRIc (>20% lifetime risk of breast cancerd) Recommend Risk-reducing salpingo-oophorectomy Discuss Option of Risk-reducing mastectomy Intervention warranted based on gene and/or risk level ATM, BRCA1, BRCA2, CDH1, CHEK2, PALB2, PTEN, STK11, TP53 BRCA1, BRCA2, Lynch syndromee BRCA1, BRCA2, CDH1, PTEN, TP53 Insufficient evidence …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nls2015/
ICARE Newsletter Summer 2015
Ask the Expert
The following question was addressed to Dr. Steven Narod who is a Tier I Canada Research Chair in Breast Cancer and a senior scientist at Women’s College Research Institute in Toronto, Canada. Dr. Narod is a world-leader in the field of breast and ovarian cancer genetics. Over the course of his career, he has profoundly shaped …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nls2015/
ICARE Newsletter Winter 2015
Highlights of the 2014 National Comprehensive Cancer Network (NCCN) Update
Genetic/Familial High-Risk Assessment: Breast and Ovarian Guidelines For breast cancer screening in BRCA carriers, yearly MRI is recommended starting at age 25; mammograms may be considered in instances where MRI is unavailable or individualized based on earliest age of onset in the family. From age 30-75, annual mammogram and breast MRI is recommended. Above age …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nlw2015/
ICARE Newsletter Winter 2015
Highlights of the 2014 National Comprehensive Cancer Network (NCCN) Update
Genetic/Familial High-Risk Assessment: Colorectal Guidelines Recommendation that tumors from newly diagnosed colorectal cancer patients be screened for Lynch syndrome (called “Universal Tumor Screening”). A new algorithm was created for Routine Tumor Testing Criteria for Lynch Syndrome Surveillance/Management recommendations were refined by gene for the various Lynch Syndrome genes. Management recommendations were refined for other inherited …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nlw2015/
ICARE Newsletter Winter 2015
PALB2: A Third Important Gene for Inherited Breast Cancer
Following the publication of an important article in the New England Journal of Medicine (NEJM) in August 2014, germline PALB2 gene mutations were confirmed as the third most important gene for inherited breast cancer, following BRCA1 and BRCA2.1 PALB2 stands for “partner and localizer of BRCA2” and is located on chromosome 16. Studies suggest that PALB2 mutations …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nlw2015/
ICARE Newsletter Winter 2015
The First PARP-Inhibitor to Be Approved for Clinical Use in BRCA Carriers
More frequently, cancer drugs are being developed to treat tumors based on their molecular make-up. PARP inhibitors are the first class of drugs specifically developed to treat BRCA-related tumors through targeting the DNA repair pathway. The PARP Inhibitors target this pathway and cause cancer cells to die while healthy cells are spared. Although PARP inhibitors …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nlw2015/
ICARE Newsletter Winter 2015
Discovery of New Colorectal Cancer Genes
New inherited cancer genes continue to be discovered with the exciting advances made possible through next-generation sequencing technologies. Recent studies identified that the POL genes predispose to inherited colorectal cancer.1,2,3 In one study, Niemenen and colleagues studied a four-generation family with Lynch Syndrome with no evidence of mismatch repair deficiency.2 Through various means (including genetic …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nlw2015/
ICARE Newsletter Winter 2015
Ask the Expert
The following question was addressed to Dr. Steven Narod who is a Tier I Canada Research Chair in Breast Cancer and a senior scientist at Women’s College Research Institute in Toronto, Canada. Dr. Narod is a world-leader in the field of breast and ovarian cancer genetics. Over the course of his career, he has profoundly shaped …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nlw2015/
ICARE Newsletter Summer 2014
Is Breast Cancer Risk Affected by Timing of Oral Contraceptives in BRCA1 Carriers?
Although oral contraceptives (OC) reduce the risk of ovarian cancer in BRCA carriers,1 it is possible that they may raise breast cancer risk. Thus it is important to understand whether age at OC use is a factor when determining impact on breast cancer risk. To address this question, a recent study (which included data from …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nls2014/
ICARE Newsletter Summer 2014
What Are Factors That Modify Cancer Risk in BRCA Carriers?
Since the discovery of the BRCA genes about two decades ago, a number of studies have reported on factors that may modify cancer risks in those who carry gene mutations. Recently, results of previously published studies were collected through a comprehensive literature review to estimate the overall effects of various risk modifiers in BRCA carriers. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nls2014/
ICARE Newsletter Summer 2014
Is There a Higher Risk of Prostate Cancer in Individuals with Lynch Syndrome?
Over the last few years, there have been studies to suggest that men with Lynch Syndrome may have a higher risk for developing prostate cancer.1,2,3,4,5 The results of these studies have differed as to whether there is an association with an aggressive form of disease. For example, some studies report the risk of developing prostate …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nls2014/
ICARE Newsletter Summer 2014
Early Results to Suggest That PSA Screening May Help to Detect Prostate Cancer Early in Men with BRCA Mutations
Over the last few years, there have been a number of studies to suggest that men with BRCA mutations, particularly BRCA2, have a higher risk of developing aggressive prostate cancer. It remains uncertain whether these men might benefit from screening through the prostate-specific antigen (PSA) test. Within the last few years, PSA screening guidelines in …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nls2014/
ICARE Newsletter Summer 2014
Ask the Expert
The following question was addressed by Dr. Steven Narod who is a Tier I Canada Research Chair in Breast Cancer and a senior scientist at Women’s College Research Institute in Toronto, Canada. Dr. Narod is a world-leader in the field of breast and ovarian cancer genetics. Over the course of his career, he has profoundly shaped …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nls2014/
ICARE Newsletter Summer 2014
New Study to Suggest Benefits of Oophorectomy in BRCA Mutation Carriers
A recent study published in the Journal of Clinical Oncology reported that prophylactic oophorectomy resulted in reducing the risk of ovarian cancer by 80%, and reduced all-cause mortality by 77%.1 The authors reported results on almost 5800 women, of whom 186 developed either ovarian, fallopian tube or peritoneal cancer. Women who had bilateral oophorectomy had …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nls2014/
ICARE Newsletter Winter 2014
Recent Evidence to Suggest That Individuals with Germline Mutations in the PTEN gene (Which Leads to Cowden Syndrome) May Have Higher Renal Cancer Risks
Cowden Syndrome is an inherited condition that leads to higher risks for breast and thyroid cancer, and possibly other cancers.1 There have been a few recent studies that suggest that this condition also puts individuals at a higher risk for kidney cancer. Specifically, Tan et al2 reported a lifetime risk of 30.6% (95% CI: 17.8-49.4) …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nlw2014/
ICARE Newsletter Winter 2014
Tamoxifen May Reduce Contralateral Breast Cancer Risk in BRCA Carriers
There have been suggestions that Tamoxifen may reduce risks for contralateral breast cancer (i.e., breast cancer in the other breast) in BRCA carriers if taken after the initial breast cancer diagnosis, based mainly on retrospective studies. Only one prospective study has looked at this question, and showed that Tamoxifen may be useful in BRCA2, but …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nlw2014/
ICARE Newsletter Winter 2014
Ask the Expert
The following question was addressed by Dr. Laronga who is a breast surgeon based at the Moffitt Cancer Center: Q. What are the risks of breast cancer after ovarian cancer in BRCA carriers? What risk management options are recommended? A. BRCA carriers remain at a higher risk of breast cancer, even after having ovarian cancer; …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nlw2014/
ICARE Newsletter Winter 2014
Contralateral Mastectomy May Improve Survival in BRCA Mutation Carriers
It has been established that women who carry a germline BRCA mutation face breast cancer risks of 60-70% in their lifetime. After an initial breast cancer diagnosis, these women face a high risk for contralateral breast cancer. Some women with BRCA mutations move forward with contralateral mastectomy when they develop their first breast cancer diagnosis (as …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nlw2014/
ICARE Newsletter Summer 2013
Sharing BRCA Test Results with Adolescent and Young Adult Children—What Does the Latest Research Show?
While there are specific recommendations against BRCA testing for minors,1 guidelines are less clear about whether parents should share their own test results with their children. Because there are no recommended surveillance or risk reduction options prior to age 25 for known BRCA mutation carriers, there has been debate about balancing the benefits of sharing …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nls2013/
ICARE Newsletter Summer 2013
US Preventive Services Task Force (USPSTF) Guidelines for Inherited Breast and Ovarian Cancer and Implications to the Affordable Care Act (ACA)
Recently, the USPSTF released updated draft guidelines in April 2013 (from those previously published in 2005) for inherited breast and ovarian cancer due to germline BRCA1 and BRCA2 gene mutations.1 USPSTF is comprised of primary care providers who review the available literature and issue guidelines about risk assessment, testing and management based on available evidence. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nls2013/
ICARE Newsletter Summer 2013
Male BRCA Carriers Have Poorer Outcomes from Prostate Cancer
Over the last few years, a number of studies have suggested that men with germline BRCA mutations (especially BRCA2) have poorer outcomes when they develop prostate cancer. In fact, a recent study of 2019 patients with prostate cancer, including 18 BRCA1 carriers, 61 BRCA2 carriers, and 1940 noncarriers indicated that germline mutations were more frequently …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nls2013/
ICARE Newsletter Summer 2013
Oophorectomy Following Menopause
Prior studies have indicated that removal of the ovaries and fallopian tubes reduces the ovarian cancer risk by ~80% and breast cancer risk by ~50%, particularly when performed pre-menopausally. However a recent case control study of 2854 pairs of women with a BRCA1 or BRCA2 mutation with or without breast cancer showed that the risk …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nls2013/
ICARE Newsletter Summer 2013
BRCA Testing: Supreme Court Update
In a landmark decision regarding the patenting of human genes on Thursday June 13, 2013, the Supreme Court of the United States unanimously ruled that human genes may not be patented. The case specifically concerned the BRCA1 and BRCA2 gene patents, held by the Utah-based company, Myriad Genetics. In the ruling, Justice Clarence Thomas wrote for the …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nls2013/
ICARE Newsletter Summer 2013
Ask the Expert
The following question was addressed by Dr. Pal, who is a Clinical Geneticist based at the Moffitt Cancer Center: Q. Does exposure to radiation increase breast cancer risk in BRCA mutation carriers? A. A number of studies have been conducted to evaluate whether BRCA mutation carriers may be more prone to radiation-induced breast cancer than …
Continue reading
Permanent link to this article: https://inheritedcancer.net/6nls2013/
ICARE Newsletter Winter 2013
Ask the Expert
The following question was addressed by Dr. Lora Thompson, a Clinical Psychologist at the Moffitt Cancer Center: Q. How do I talk to family members about my genetic test results? A. The ability to share risk information with family members is a common reason why many individuals undergo genetic testing. Family members may feel appreciative …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nlw2013/
ICARE Newsletter Winter 2013
Points to Consider Regarding Bilateral Salpingectomy as a Risk Reduction Procedure for Ovarian Cancer
Over the last few years, there has been evidence to suggest that a substantial proportion of ovarian cancer may start in the fallopian tubes, although some cancer clearly arises in the ovary. As a result, removal of both fallopian tubes (called ‘bilateral salpingectomy’) has been suggested as an interim procedure to reduce risk in BRCA …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nlw2013/
ICARE Newsletter Winter 2013
The Selection of Chemotherapy in BRCA Patients with Pancreatic Cancer
Some evidence suggests that individuals with BRCA mutations who develop pancreatic cancer may benefit from specific chemotherapy regimens. In a recent review of this topic, Kim et al reported on a study of 5 patients with BRCA mutations (4 BRCA2 and 1 BRCA1) who were treated with a platinum-based chemotherapy regimen.1 Of these patients, 3 …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nlw2013/
ICARE Newsletter Winter 2013
Is Lynch Syndrome Associated with Breast Cancer?
The cancer spectrum typically seen in individuals with Lynch Syndrome includes cancers of the colon, endometrium, ovary, stomach, and other cancers (including cancer of the renal pelvis, ureter, small bowel and pancreas). The issue of whether breast cancer risk is elevated in those with Lynch syndrome has been controversial, with conflicting results between various studies. …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nlw2013/
ICARE Newsletter Summer 2012
Ask the Expert: What are the risks of hormone replacement therapy (HRT) on breast cancer risk in women with BRCA mutations?
The following questions were addressed by Drs. Pal and Lancaster at the Moffitt Cancer Center: Q. What are the risks of hormone replacement therapy (HRT) on breast cancer risk in women with BRCA mutations? A. Concern about HRT in BRCA carriers is its potential to raise the risk of breast cancer, as seen in the …
Continue reading
Permanent link to this article: https://inheritedcancer.net/4nls2012/
ICARE Newsletter Summer 2012
New Study Suggests Breast Cancer Risk for Non-Carriers of Family-Specific BRCA Mutations Is Not Increased
Data from a population-based study was recently reported to estimate breast cancer risk among family members who tested negative for a known BRCA1/2 family mutation (i.e., non-carriers). The study included 3047 women diagnosed with breast cancer. Results from the study indicated there was no increased risk for breast cancer in non-carriers as compared to family …
Continue reading
Permanent link to this article: https://inheritedcancer.net/3nls2012/
ICARE Newsletter Summer 2012
Emerging Cancer Panels for Testing Patients for Inherited Cancer Predisposition
Genetic testing for inherited cancer predisposition is typically performed by testing for one condition at a time. However, with the tremendous advances in genetic testing technologies over the last few years, the cost of testing has plummeted. To put this into perspective, the first human genome cost 2-3 billion dollars to sequence and took over …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nls2012/
ICARE Newsletter Summer 2012
Ask the Expert: What are the recommendations for screening following prophylactic surgeries?
The following questions were addressed by Drs. Pal and Lancaster at the Moffitt Cancer Center: Q. What are the recommendations for screening following prophylactic surgeries? A. In BRCA mutation carriers, bilateral prophylactic mastectomy reduces the risk of breast cancer by over 90%. It essentially lowers the risk of breast cancer to below that of the …
Continue reading
Permanent link to this article: https://inheritedcancer.net/5nls2012/
ICARE Newsletter Summer 2012
Prostate Cancer Screening Recommendations for Men with BRCA Mutations
Over the last few years, there have been several studies that suggest that men with BRCA mutations are at a higher risk for developing and dying from aggressive prostate cancer. It is possible that PSA testing may be of benefit in men with BRCA mutations. However, until the utility of PSA is determined in these …
Continue reading
Permanent link to this article: https://inheritedcancer.net/2nls2012/
ICARE Newsletter Summer 2011
Ask the Expert
We are fortunate to have Dr. Alvaro Monteiro, who is a molecular geneticist and expert on the BRCA1 and BRCA2 genes, as a member of our team. For our first ICARE newsletter, Dr. Monteiro teaches us about Variants of Uncertain Significance. Q. What is a Variant of Uncertain Significance (VUS) test result? How do researchers …
Continue reading
Permanent link to this article: https://inheritedcancer.net/1nls2011/
")